
Abstract
The S=O stretching and SOH bending peaks in the vibrational spectra of HSO4−(H2O) n , with n up to 6, are analyzed by both harmonic analysis and ab initio molecular dynamics simulation. The SOH bending mode is found to be much more sensitive to the extent of hydration and to the fluctuation of hydrogen bonds than the S=O stretching mode. The SOH donor hydrogen bond is gradually stabilized by n = 4, and further shortened up to n = 6, which is the key factor to understand the trend of evolution observed in the infrared multiple photon dissociation spectra.
Classification dependents by the distance of hydrogen bond.

Introduction
Bisulfate ion is one of the trace sulfur species in the atmosphere, in addition to H2SO4, SO42−, and their precursor SO2 from both natural and anthropogenic sources.1,2 Although not as abundant as its neutral cousins, HSO4− could play an important role in aerosol formation, and its interaction with water molecules is one of the important factors underlying the homogeneous nucleation processes in the atmosphere.3–8 Hydrated bisulfate cluster ions, HSO4−(H2O) n , are molecular models to probe such interactions and the subject of a number of experimental and theoretical studies. Theoretical calculations have identified many local energy minimum structures,9–12 with varying harmonic frequencies and hydration energy. In the 600–1800 cm−1 region, the vibrational profiles of HSO4−(H2O) n have been measured by infrared multiple photon dissociation (IRMPD) spectroscopy, with n up to 16, and assigned by comparison with the harmonic spectra obtained for optimized structures. 13 More recently, the photoelectron spectra for HSO4−(H2O) n , with n up to 2, have also been reported. 14
Bisulfate ion is also one of the conjugate bases, ubiquitous in aqueous solution, and often encountered in general chemistry lab. Micro-solvation details around such polyatomic ions are of fundamental interests, and recent spectroscopic studies have shed considerable lights into the geometry, electronic structures, and dynamics in such hydrated cluster ions.15–23 Particularly relevant to HSO4−(H2O) n are the recent studies on H2PO4−(H2O) n and HCO3−(H2O) n .24–28 All three singly charged anions have two kinds of groups, X=O and XOH, with the XO stretching (υ(X=O)) and the XOH bending (δ(XOH)) coupled together with considerable overlap. Furthermore, δ(XOH) is more sensitive than υ(X=O) to the fluctuation in hydrogen bond (HB) distances. As the number of water molecules increases and the XOH group becomes better solvated, it would produce a trend of changes in the pattern of υ(X=O) and δ(XOH) peaks, which could be explained by the dynamic of HB fluctuation.26,28
Such dynamics is also expected to be important for HSO4−(H2O) n . Moreover, with a pKa value of 2.0, HSO4− is more acidic than H2PO4−and HCO3−, and the acidic dissociation of HSO4−, when the number of water molecules increases, has been reported by a recent research. 29 In this paper, we report ab initio molecular dynamics (AIMD) simulation results on HSO4−(H2O) n , with n up to 6, and show that the solvation of SOH is also the key to understand the trend of change in the υ(S=O) and δ(SOH) peaks with increasing cluster size, as observed in the IRMPD experiment. 13
Computational methods
The optimization of 0 K structures and harmonic analysis are performed by GAUSSIAN 16 package (Rev A.03) at the B3LYP/6-311+G(d,p) level.30–32 Relative energies include zero-point vibrational energies. A scaling factor of 1.071 is used for low-frequency region below 2000 cm−1. 33 The resulting stick spectra are convoluted by a Gaussian line shape function with a full width at half-maximum line width of 15 cm−1.
AIMD simulations at finite temperatures are performed by the CP2K package. 34 A cluster under simulation is put at the center of a periodic cubic box with a length of more than twice the largest interatomic distance in the cluster and with the effect of the periodical charge density images eliminated by the decoupling technique developed by Martyna and Tuckerman. 35 At every time step, the potential energy and atomic forces are calculated within the framework for density functional theory (DFT). The wave functions are represented by double-zeta Gaussians, and the electron density is represented using a mixed basis set of Gaussians and auxiliary plane waves with a cut-off of 320 Rydberg. The Goedecker–Teter–Hutter (GTH) typed pseudopotentials are employed for the atomic cores. For the exchange and correlation energy, BLYP functional are employed with dispersion corrected by Grimme’s D3 scheme. 36
The atomic motion is treated by Newtonian mechanics, with the temperature controlled by a Nose–Hoover thermostat and with a time step of 0.5 fs. The temperature is first scaled at the desired value for 3 ps, followed by a data collection run in the NVE (micro-canonical ensemble) ensemble for 15 ps. The value of dipole moment is calculated and collected at each time step, so that a vibrational spectrum can be directly simulated by the Fourier transform of the dipole time-correlation function (DTCF)
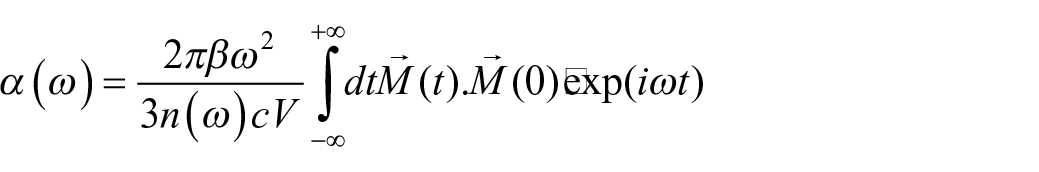
where
Results and discussion
HB fluctuation in HSO4−(H2O)
In Figure 1, three optimized structures are shown. The energy gap among them is less than 2 kJ mol−1.

Optimized structures of HSO4−(H2O). Relative energy in parenthesis is in kJ mol−1.
In AIMD simulations, these two donor HBs in
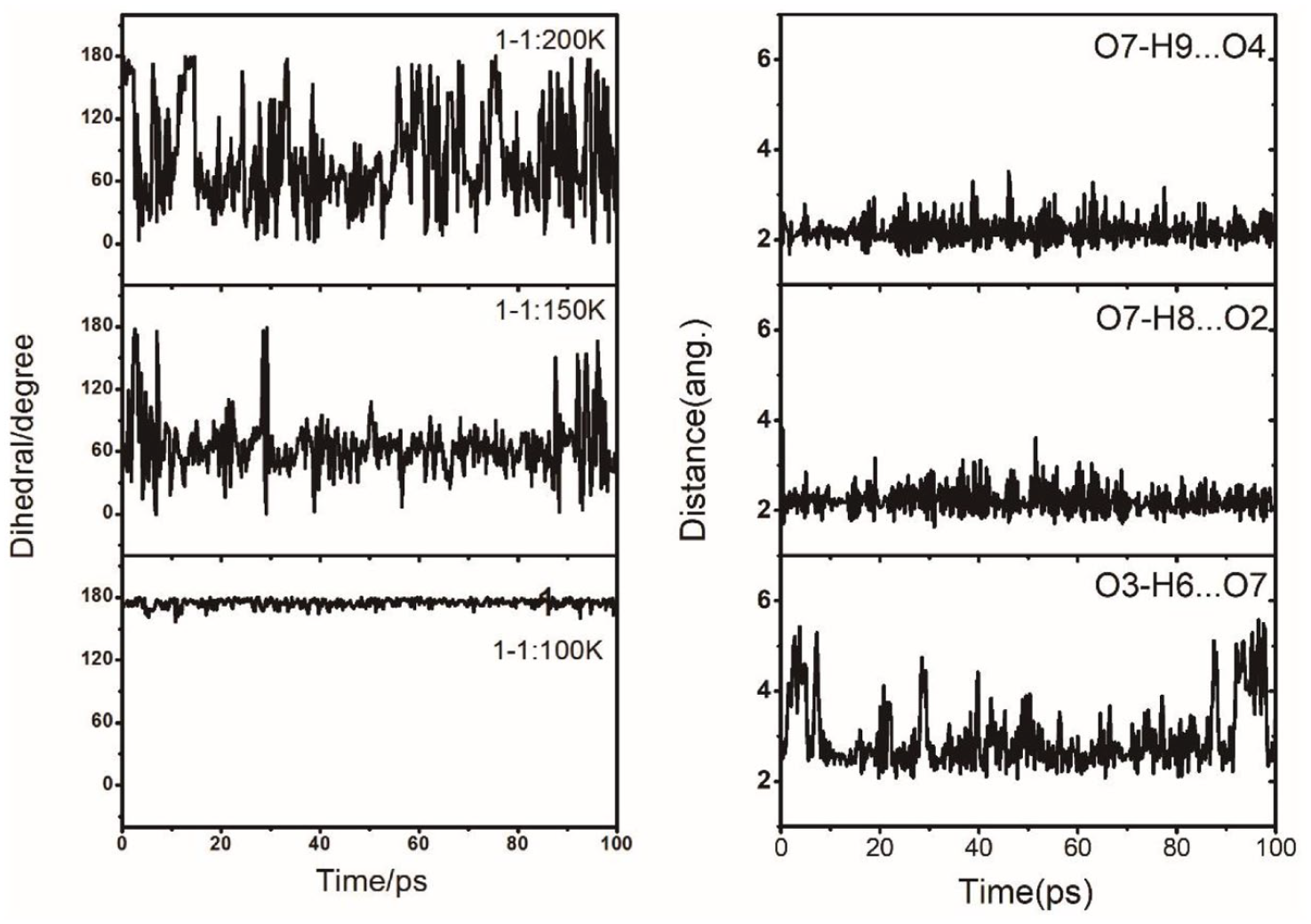
Left panel: fluctuation in the dihedral-O5S1O3H6 during AIMD simulations for cluster
The dynamics of HSO4−(H2O) at finite temperature is dominated by the transformation between
S=O stretching and SOH bending in HSO4−(H2O)
Although SOH···OH2 is a weak HB with a distance ~2.5 Å, it has a significant impact on the vibrational profile between 1000 and 1400 cm−1. There are four peaks in this region: the symmetric stretching of the three S=O bonds, υS(S=O); the asymmetric stretching of the two S=O bonds, cis to the HSO group, υAS-C(S=O); the asymmetric stretching of the S=O bond, trans to the SOH group, υAS-T(S=O); and finally, the bending of S-O-H, δ(SOH). For the naked HSO4−, the frequency ordering is

and there are couplings between δ(SOH) and υAS-T(S=O). Upon solvation by one H2O, when the SOH···OH2 HB is not formed as in
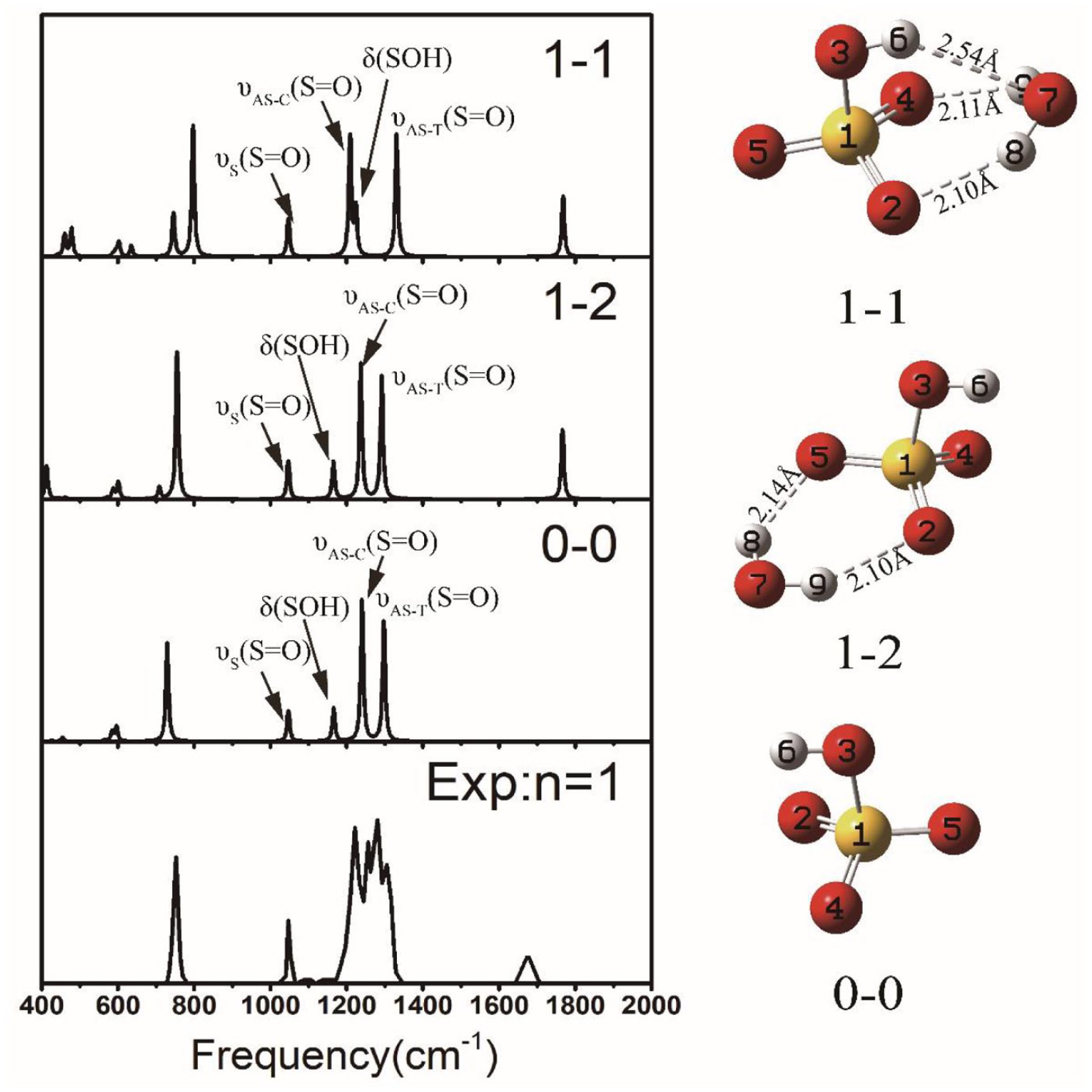
Harmonic spectra for naked HSO4−, clusters
The DTCF spectra for

Harmonic and DTCF spectra for
The effects of SOH solvation on SOH bending and S=O stretching
The strength of SOH···OH2 HB also provides the key to understand the evolution of spectral region between 1000 and 1400 cm−1. In my work, I did some classification and quantification research by the distance of this HB: naked SOH with infinite HB distance; solvated SOH with HB distance above 2 Å; and solvated SOH with HB distance under 2 Å. Below n ⩽ 4, a number of clusters have been optimized with the SOH group being unsolvated, and harmonic spectra are shown in Figure 5. The S=O groups become more solvated with more water molecules, and meanwhile the inter-water HB network becomes more elaborate. But such changes do not have any bearings on the pattern of the δ(SOH) and υ(S=O) peaks. As long as SOH is unsolvated, the patterns of δ(SOH) and υ(S=O) peaks are all similar to those for unsolvated HSO4−, with the same frequency of ordering


Optimized structures of HSO4−(H2O)n, n = 0–4, with the SOH group being unsolvated. A scaling factor of 1.071 is used in the harmonic spectra for this region.
δ(SOH) is 1160–1166 cm−1 and υ(S=O) is 1045–1047 cm−1 for all these clusters with the unsolvated SOH group. It means the position of vibrational modes will not change with the size evolution.
Figure 6 shows three structures in which the SOH group is solvated by a typical donor HB with a distance above 2 Å. The acceptor H2O, while forming two donor HB with S=O groups, is not solvated further by any other water molecule. Their spectra between 1000 and 1400 cm−1 are similar, and the frequency ordering is switched to


Optimized structures of HSO4−(H2O)n, n = 1–3, with the SOH group being solvated by an HB with a distance above 2 Å. A scaling factor of 1.071 is used in the harmonic spectra for this region.
δ(SOH) is 1224 cm−1 for
Starting at n = 2, the water molecule accepting SOH in hydrogen bonding could form donator HB with other water molecules. For


Optimized structures of HSO4−(H2O) n , n = 2–6, with the SOH group being solvated by an HB with a distance below 2 Å. A scaling factor of 1.071 is used in the harmonic spectra for this region.
δ(SOH) is 1406 cm−1 for
Furthermore, δ(SOH) is decoupled from the υ(S=O) modes, in contrast to the previous two types of structures, for which there is considerable coupling between δ(SOH) and υAS-T(S=O). When the water molecule solvating an XOH group is connected to other water molecules by HBs, the impact of δ(SOH) is absorbed by surrounding water molecules and lose its effects on S=O stretching. Such decoupling between XOH bending and X=O stretching has been observed in the cases of H2PO4−(H2O) n and HCO3−(H2O) n .26,28
For
The evolution of SOH bending and S=O stretching with cluster size
With the effects of SOH solvation understood, it is more straightforward to assign the region between 1000 and 1400 cm−1 for the larger clusters. The SOH group is well solvated for n ⩾ 5. As shown in Figure 8, peaks E, D, and C are due to the three S=O stretching modes, υS(S=O), υAS-C(S=O), and υAS-T(S=O), respectively, in agreement with the previous assignment.
13
Higher in frequency than these three peaks is peak B, due to δ(SOH), as the HB of SOH···OH2 is strengthened by such cluster sizes. The three υ(S=O) peaks (C, E, and D) are less sensitive to n value increases. Since these modes do not directly involve the movement of H atoms. It is perfectly reproduced by both harmonic analysis and DTCF spectra obtained by AIMD simulations shown in Figure 8. Due to the overestimate of HB strength, the calculated gap between peak B/δ(SOH) and other υ(S=O) peaks in the DTCF spectrum is 1450 cm−1 to 1046 cm−1 for

DTCF spectra for
The overall pattern for these four peaks changes little as n increases from 9 to 11, except that the height of peak B/δ(SOH) drops appreciably. The flattening of the δ(XOH) with increasing n peak has been observed in the cases of H2PO4−(H2O)
n
and HCO3−(H2O)
n
.26,28 It can be again attributed to the strengthening of the XOH···OH2. As shown in Figure 9,
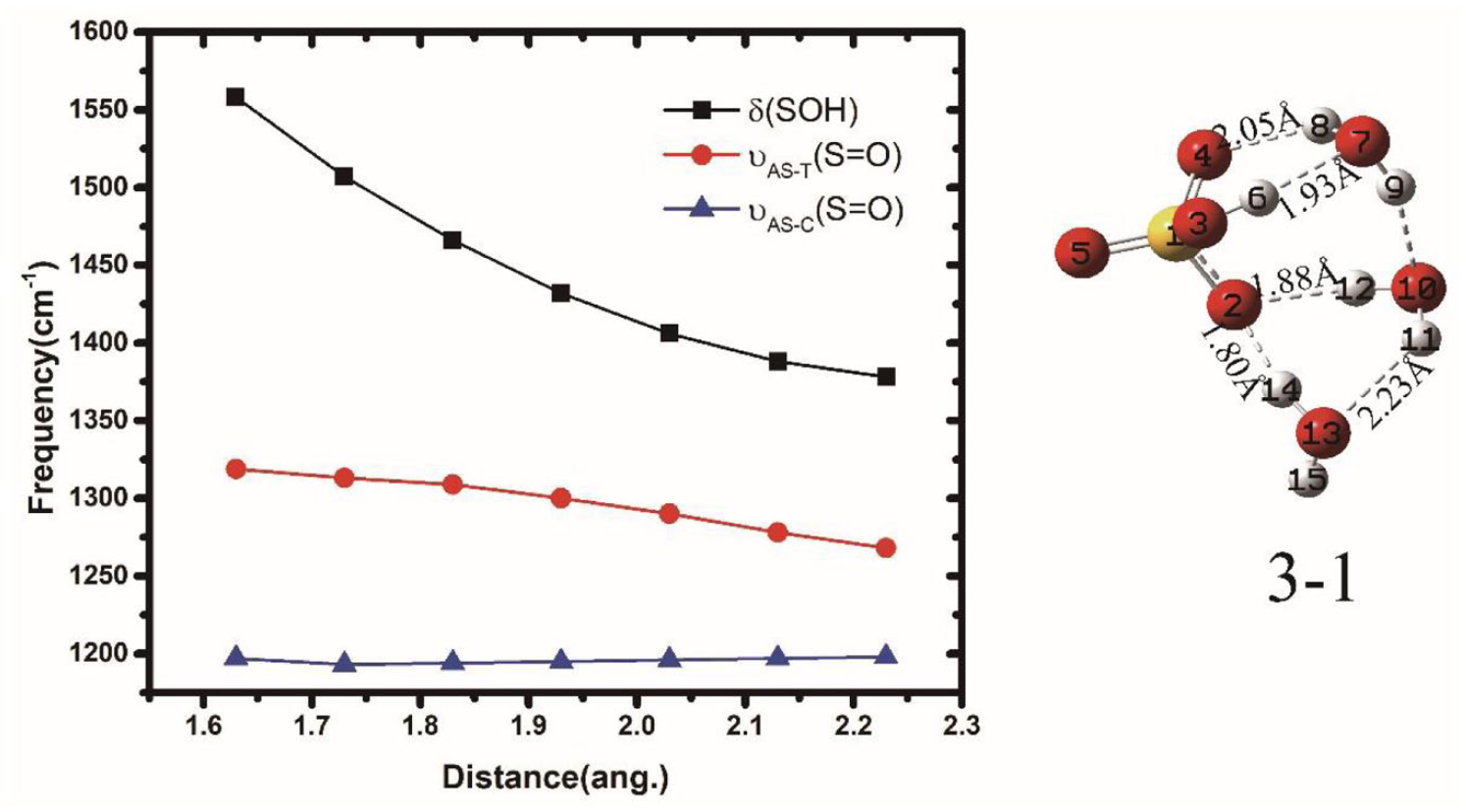
Simulated harmonic frequency of HB SO-H6···O7 for
The spectral assignment for the smaller cluster with n = 2−4 is more complicated. For example, the SOH···OH2 distance for the optimized

Harmonic, DTCF, and experimental IRMPD spectra for
The evolution of SOH in larger cluster size
In our previous study, 29 we found that although bisulfate anion is a weak acid with a pKa of 2.0, HSO4−(H2O) n is completely dissociated at a moderate size of n = 16 and also dissociates in the liquid environment. For such charged clusters, the size can be precisely selected, making it possible to probe the molecular details about the acidic dissociation from its onset around n = 12 to its completion around n = 16. However, in the small cluster size of n = 1–6, the SOH group remains without dissociation.
Conclusion
In summary, the evolution of the IRMPD spectra for HSO4−(H2O) n versus the cluster size can be attributed to the solvation of SOH group. For n ⩽ 4, SOH is not well solvated, which is indicated by a relative strong peak B, due to υAS-T(S=O). There is a significant coupling between υAS(S=O) and δ(SOH) peaks. For 5 ⩽ n ⩽ 6, SOH is well solvated, with a donor HB shorter than 2 Å. The spectral pattern is υS(S=O) < υAS-C(S=O) < υAS-T(S=O) < δ(SOH), with δ(SOH) decoupled from υAS(S=O) and broadened. It can be concluded that the SOH donor HB is the key factor to understand the trend of evolution observed in IRMPD spectra.
Footnotes
Acknowledgements
The author thanks to her supervisor Professor Liu Zhifeng of The Chinese University of Hong Kong.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Jiangsu (grant 20KJB150033), the Jiangsu Innovative Research Program for Talent from the World’s Famous Universities, and the Jiangsu Second Normal University (JSNU927301).