
Abstract
Urolithins are the gut microbiota metabolites of ellagitannins which are found in natural plants such as pomegranate, strawberry, and raspberry, and in nuts. Recently, several reports have clarified the underlying mechanism of urolithins in central nervous system inflammation. Therefore, urolithins have become potential therapeutic drug candidate molecules for central nervous system diseases. Derivatives

Introduction
Ellagitannins (ETs) and ellagic acid (EA) are polyphenolic compounds that are found in a variety of plants, such as pomegranate, raspberries, and in nuts.1,2 Multiple studies have shown that foods that are rich in ETs or EA have potential in improving neurodegenerative diseases and brain inflammation. For example, the release of proinflammatory cytokines in the brains of mice with Alzheimer’s disease (AD) can be inhibited by dietary supplementation of pomegranate. 3 Processing of the amyloid beta precursor protein in aging transgenic mice can be modulated by short-term use of pomegranate extract (containing 68.2% ET and 2.3% EA). 4 Improved cognitive and motor performance in aged rats that have been fed with walnuts that contain ETs has also been treported. 5 However, the applications of ETs and EA are limited due to their direct absorption and difficulties with utilization in vivo in the field of medicines and health products.
ETs and EA can be metabolized to urolithins (6H-dibenzo[b,d]pyran-6-one derivatives), and persist in the enterohepatic circulation.6–9 In vivo, urolithins may have biological activities related to ETs and EA. Among them, urolithin A (UA) and urolithin B (UB) are observed metabolites,10–12 which have been reported to demonstrate biological activities, such as regulation of estrogen and androgen receptors; anti-oxidant, anti-inflammatory, anti-aging, anti-cancer, and anti-diabetes activities; and so on.13–16 Some recent studies have shown that UA and UB can produce neuroprotective effects by reducing LPS-induced BV2 microglial inflammation, through inhibition of the NF-κB, MAPK (p38 and Erk1/2), and Akt signaling pathways. 17 Other research groups have found that the neuroprotective effect of pomegranate on AD is mediated by UA and UB and their methyl derivatives methoxy-urolithin A (mUA) and methoxy-urolithin B (mUB). 2 Thus, urolithins have great potential for neuroprotection and improving neurodegenerative diseases and brain inflammation.
In recent years, phosphodiesterase 2 inhibitors have attracted much attention due to their potential therapeutic effects in central nervous system diseases. The single gene-encoded phosphodiesterase PDE2 is mainly distributed in the brain, the heart, and in neurons. 18 With PDE2 as a potential drug target, it plays an important role in maintaining the levels of cAMP and cGMP, and its selective inhibition was expected to be useful in improving endothelial permeability and memory, and to treat central nervous system diseases such as AD.19–21
BAY60-7550 is an important PDE2 inhibitor that was discovered recently. Studies have reported its efficacy in enhancing learning, memory, and cognitive function. 22 Zhu has reported the X-ray crystal structure (4HTX) with the complex formed by PDE2 and a highly selective inhibitor (BAY60-7550) 23 that provided a basis for the design of new PDE2 inhibitors, yet there are no efficient PDE2 inhibitors on the market. Therefore, there is an urgent clinical need to develop novel PDE2 inhibitors.
In this study, a series of urolithin derivatives has been designed by combining the structures of UA, UB, and mUA based on the basic principles of drug design. 24 The relationship between small molecule compounds and the PDE2 protein crystal 4HTX was simulated by Discovery Studio (DS) software. All compounds were screened by the structure–activity relationship score, which is the basis for their synthesis. Finally, the inhibitory effect of the compounds on PDE2 was verified at the enzyme level, and the structure–activity relationship was analyzed. The structures of UA, UB, and mUA are shown in Figure 1.

Chemical structures of UA, UB, and mUA.
Results and discussion
Synthetic pathways
In previous studies, the parent urolithin nucleus was targeted. As described in Figure 2, in this study, we aimed at designing and synthesizing derivatives of 4-hydroxybiphenyl, 4-hydroxy-4′-methoxybiphenyl and 4,4′-dihydroxybiphenyl; derivatives of 3-hydroxyphenyl benzoate; and 3-hydroxyphenyl 3-methoxybenzoate, together with derivatives of N-(3-hydroxyphenyl)benzamide, N-(3-hydroxyphenyl)-3-hydroxybenzamide and N-(3-hydroxyphenyl)-3-methoxybenzamide by engineering the parent nuclei of UA, UB. and mUA.

The modification of UA, UB, and mUA described in this work.
The synthetic methods used to prepare the biphenyl series of derivatives is shown in Scheme 1. 4-Hydroxybiphenyl, 4-hydroxy-4’-methoxybiphenyl or 4,4′-dihydroxybiphenyl, K2CO3 and R1X were reacted to generate the desired biphenyl series of derivatives in good yields. The synthesis of the benzoate derivatives is shown in Scheme 2 and comprised three steps: First, resorcinol was alkylated using K2CO3 and R1X. Second, the benzoyl chloride was prepared, and, finally, the acylation of the alkylated resorcinol was carried out. The synthesis of the benzamide derivatives was accomplished using similar procedures and is shown in Scheme 3. The isolation of O-alkylation products from 3-hydroxyaniline is of note. This has literature precedent but, as the yields were only modest, may also be due to selective extraction of the O-alkylated product from the basic aqueous solution during work up, the deprotonated N-alkylated material remaining in the aqueous solution; see experimental.

Synthesis of a series of derivatives of 4-hydroxybiphenyl (

Synthesis of a series of derivatives of 3-hydroxyphenyl benzoate (

Synthesis of a series of derivatives of N-(3-hydroxyphenyl) benzamide (
Molecular docking results
In order to screen out the compounds with high affinity to a specific receptor binding region of the PDE2 protein, and to verify whether the designed compounds have a good binding effect with the PDE2 protein, we used the CDOCKER module of DS to simulate the docking results of the PDE2 protein and the designed compounds. All docking results indicated that for the compounds, a higher –CDOCKER–INTERACTION–ENERGY level predicted a higher affinity for the PDE2 protein. Meanwhile, we also verified the affinity of BAY60-7550 and the PDE2 protein, 25 and its –CDOCKER–INTERACTION–ENERGY value was 57.95.
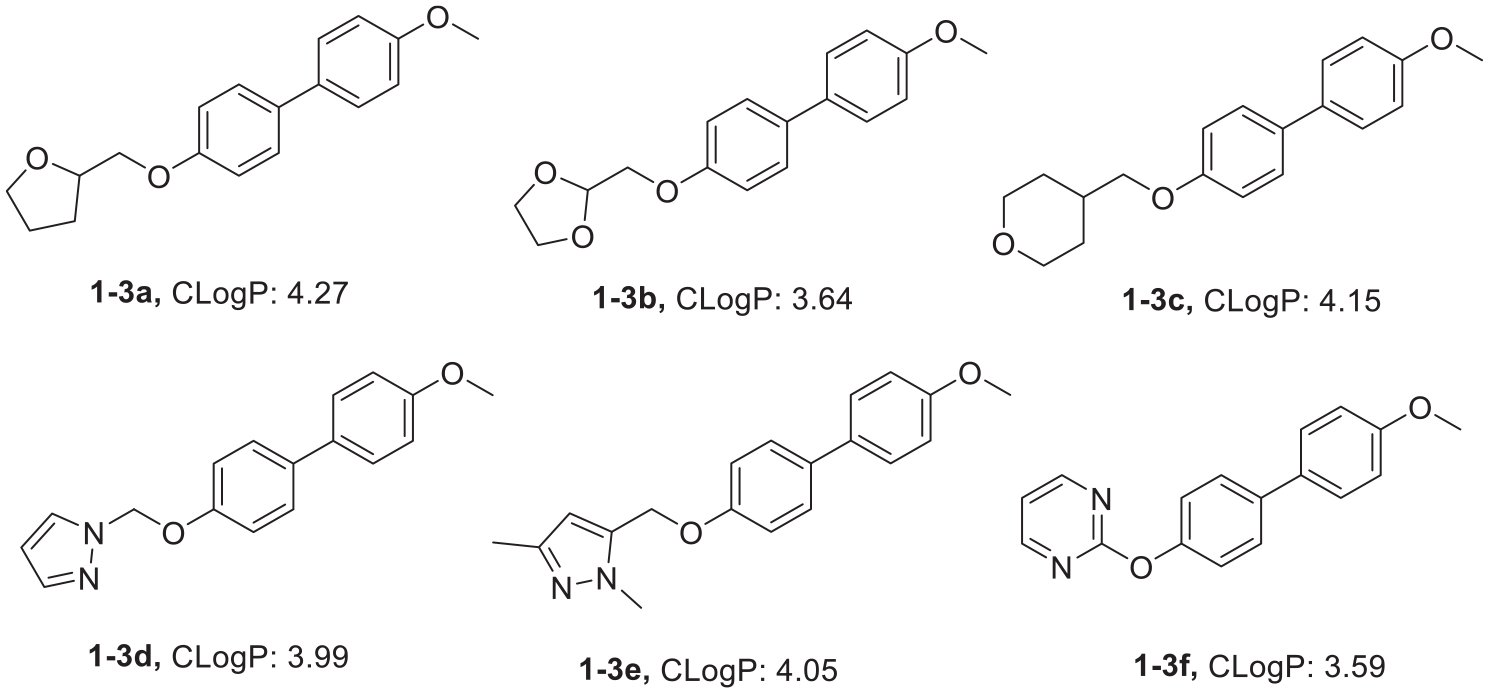
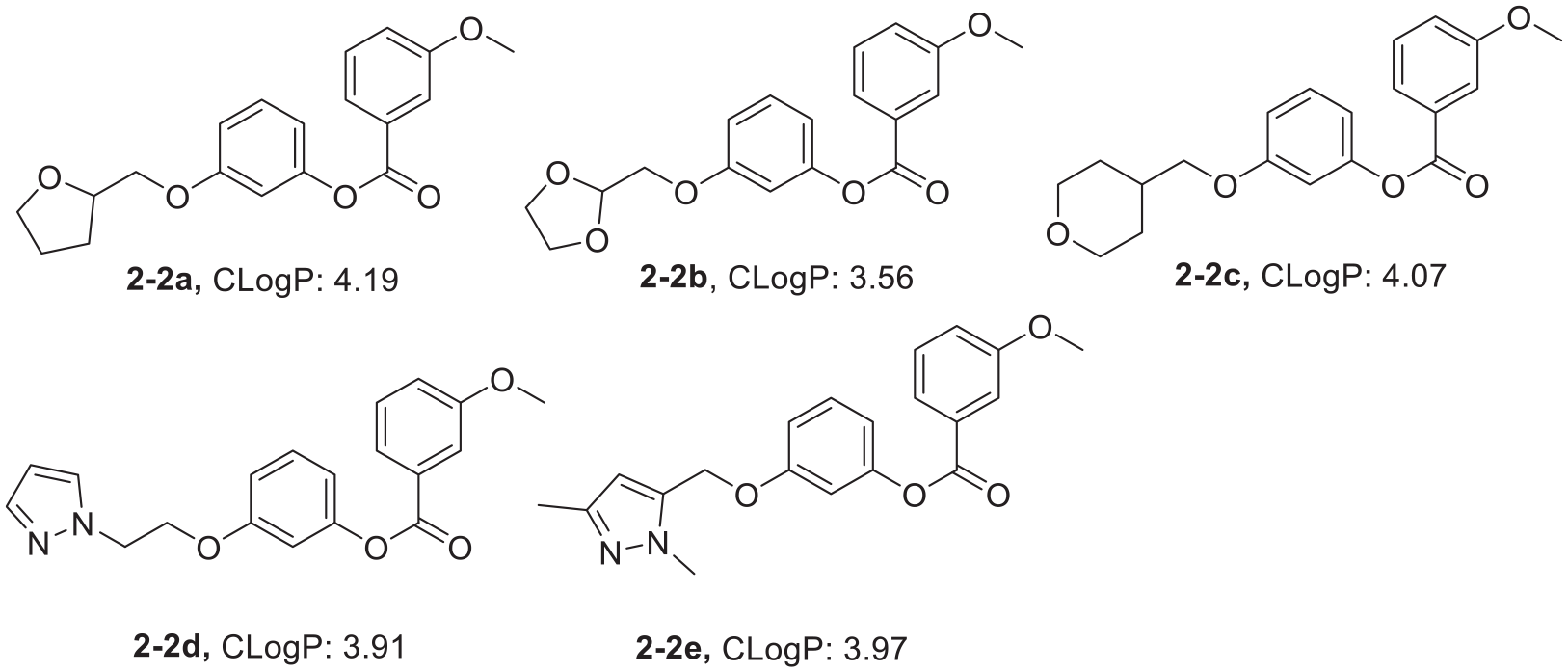
Moreover, the five basic rules for screening drugs were referred into the designed derivatives, namely, the rule of 5 (Ro5), for which the molecular weight is less than 500, the number of hydrogen-bonded donors (including hydroxy, amino, etc.) does not exceed five, the number of hydrogen-bonded receptors does not exceed 10, and the logarithmic value (CLogP) of the theoretical oil–water partition coefficient of the compound is not more than 5, and the number of rotatable keys does not exceed 10. The groups for the etherification reaction we designed are mainly C2–C9 alkyl, cycloalkyl-substituted C1–C9 alkyl, heterocyclyl-substituted C1–C9 alkyl, and aryl-substituted C1–C9 alkyl and C2–C9 heterocyclic bases. From the obtained docking results, the compounds have higher docking scores (Tables 1–3) when the etherified groups are C5–C7 substituted alkyl, cycloalkyl, aryl, and heterocyclic substituents. This can probably be explained by the fact that when the substituent is a group composed of 5–7 carbons, which can just occupy the pocket in protein crystal, the groups can induce the amino acid residues which in the protein crystal pocket to generate hydrogen bonding forces with small molecule compounds to produce good docking results. When the substituent contains a smaller number of carbons (C1–C4), the group cannot completely occupy the pocket of the protein crystal and produces more hydrogen bonding forces, which makes the docking results unsatisfactory. However, when the substituent contains more carbons (C8–C9), due to the limited space in the pocket of the protein crystal, it seldom completely contains the small molecule compounds, so that part of the structure extends to the outside of the pocket, which leads to a failure in generating hydrogen bonding forces and a failure to obtain good docking results. In conclusion, the docking effects of our designed derivatives are better than that of the lead compounds UA, UB, mUA, indicating that the designed derivatives are better than the lead compounds in terms of affinity. Hence, these data provide theoretical support for the synthesis of the designed series of derivatives.
Docking results of the biphenyl series of compounds with 4HTX.
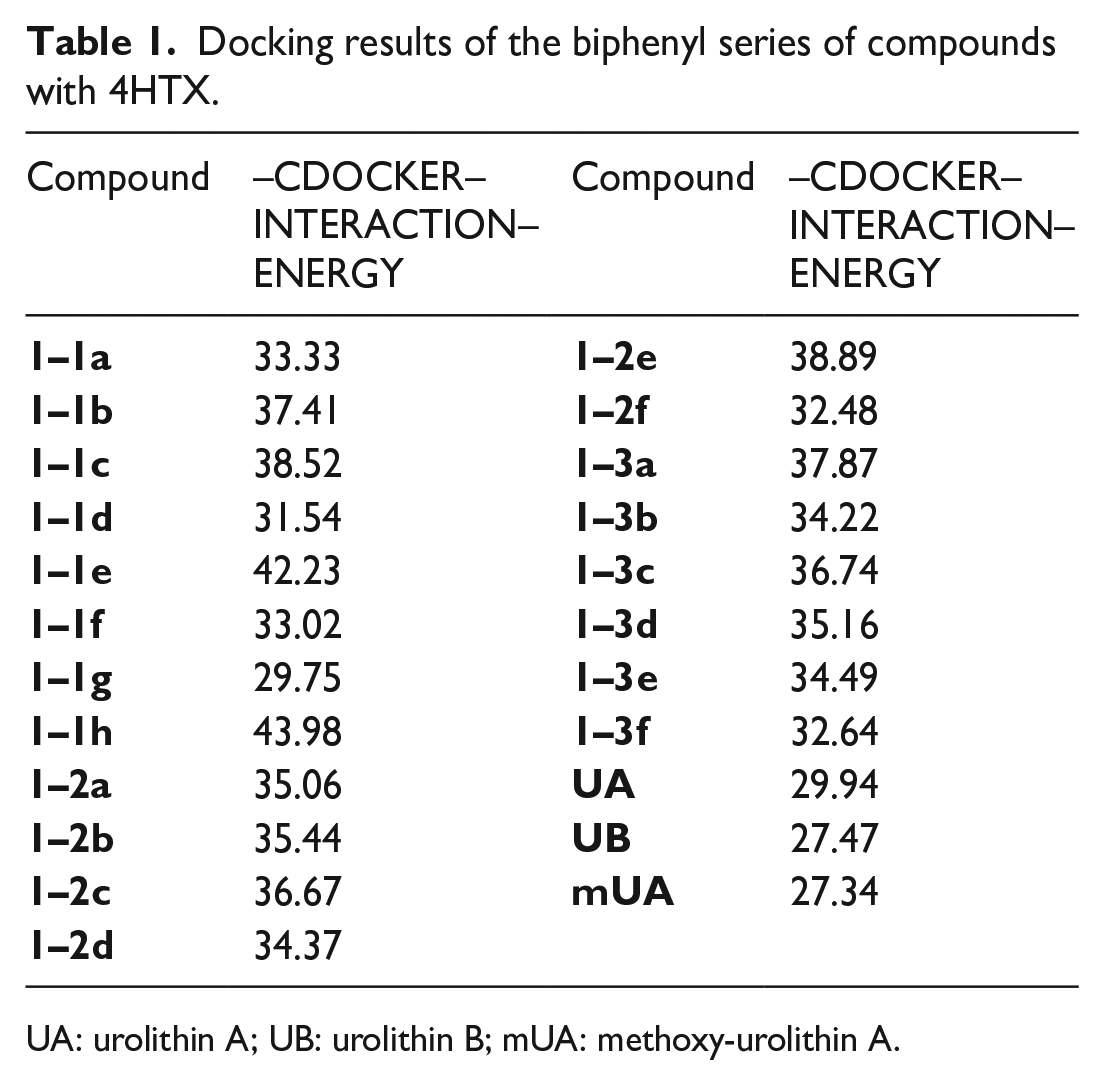
UA: urolithin A; UB: urolithin B; mUA: methoxy-urolithin A.
Docking results of the benzoate compounds with 4HTX.

UA: urolithin A; UB: urolithin B; mUA: methoxy-urolithin A.
Docking results of the benzamide compounds with 4HTX.
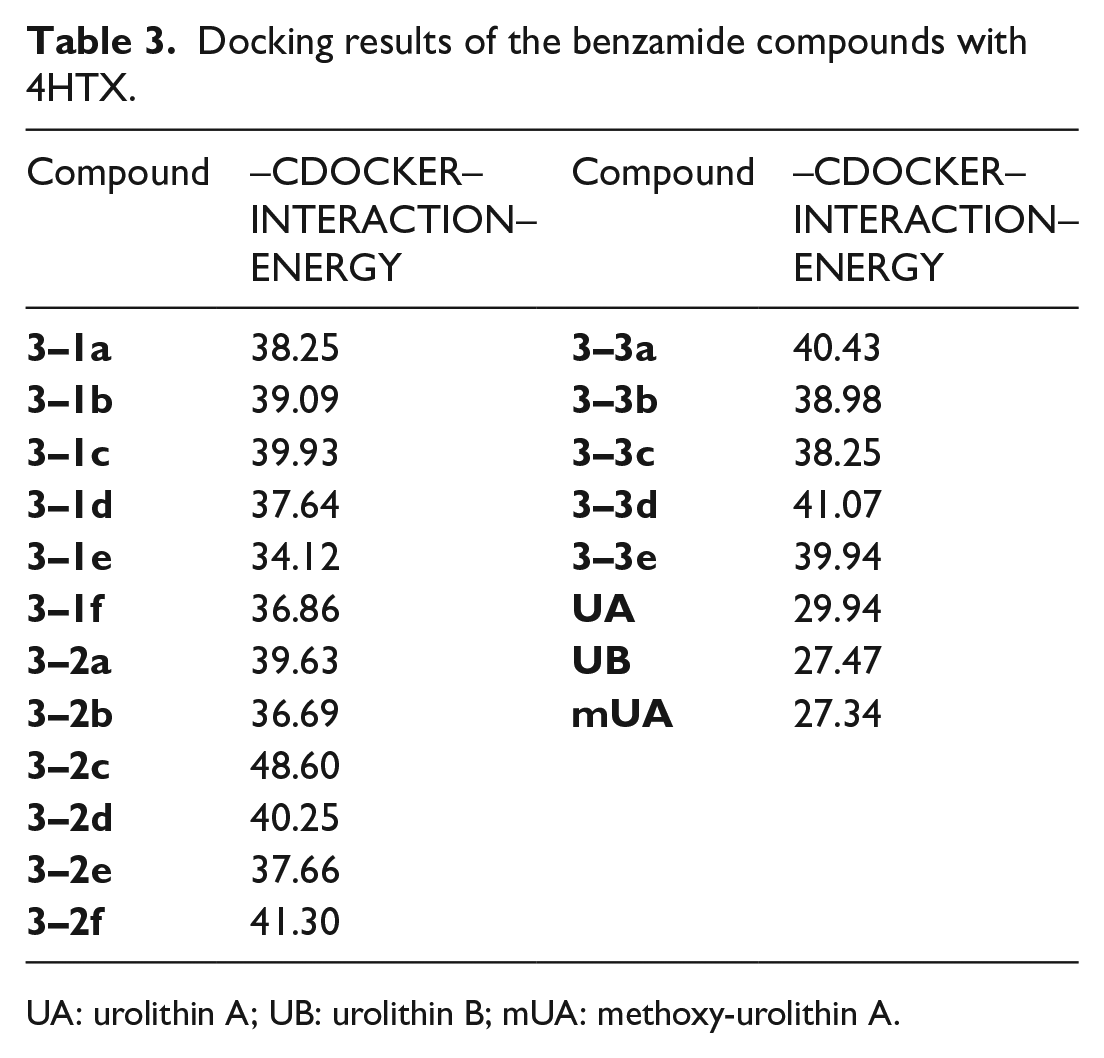
UA: urolithin A; UB: urolithin B; mUA: methoxy-urolithin A.
BAY60-7550 is an important nanomolar PDE2 inhibitor that was recently discovered. The reasons for its high selectivity and inhibition have been explained by analyzing its interactions with the crystals of the catalytic domain of PDE2A. 23 BAY60-7550 mainly binds to two subbags of the active site: the Q pocket containing a glutamine switch and a hydrophobic clip, and the H pocket containing hydrophobic amino acid residues. BAY60-7550 mainly occupies the Q pocket, and the glutamine switching mechanism is also used for binding with the active site, that is, conservative residues Gln859 and Gln812 are used as purine binding sites, and Phe862 in the hydrophobic clamp forms parallel π–π stacking interactions. In the H pocket, the hydrophobic propylphenyl of BAY60-7550 also binds to hydrophobic residues such as Leu770, Leu809, Ile866, Ile870, and so on. It was thus docked with 4HTX (crystal structure of PDE2A catalytic domain complex) in the PDB database by using molecular docking software, and the interaction forces are shown in Figure 3.

Binding mode of BAY60-7550 within the active site pocket of human PDE2 (PDB ID:4HTX).
Compound

Binding mode of
Enzymatic assays
As shown in Figures 5–12, most of the derivatives we designed have appropriate CLogP values (2.0–5.0), which implies that they have good blood–brain barrier penetration. AlphaScreen kit was used to detect the PDE2 inhibitory activity of the synthetic derivatives in vitro. Each compound was diluted at seven concentrations, and three groups of parallel experiments were carried out for each group. The IC50 value was calculated by fitting with Graphpad Prism 7 software. The results (Table 4) show that, compounds

CLogP values of the 4-hydroxybiphenyl derivatives.

CLogP values of the 4,4′-dihydroxybiphenyl derivatives.
CLogP values of the 4-hydroxy-4′-methoxybiphenyl derivatives.

CLogP values of the phenyl 3-methoxybenzoate derivatives.
CLogP values of the 3-hydroxyphenyl-3-methoxybenzoate derivatives.

CLogP values of the N-(3-hydroxyphenyl)benzamide derivatives.

CLogP values of the 3-hydroxy-N-phenylbenzamide derivatives.
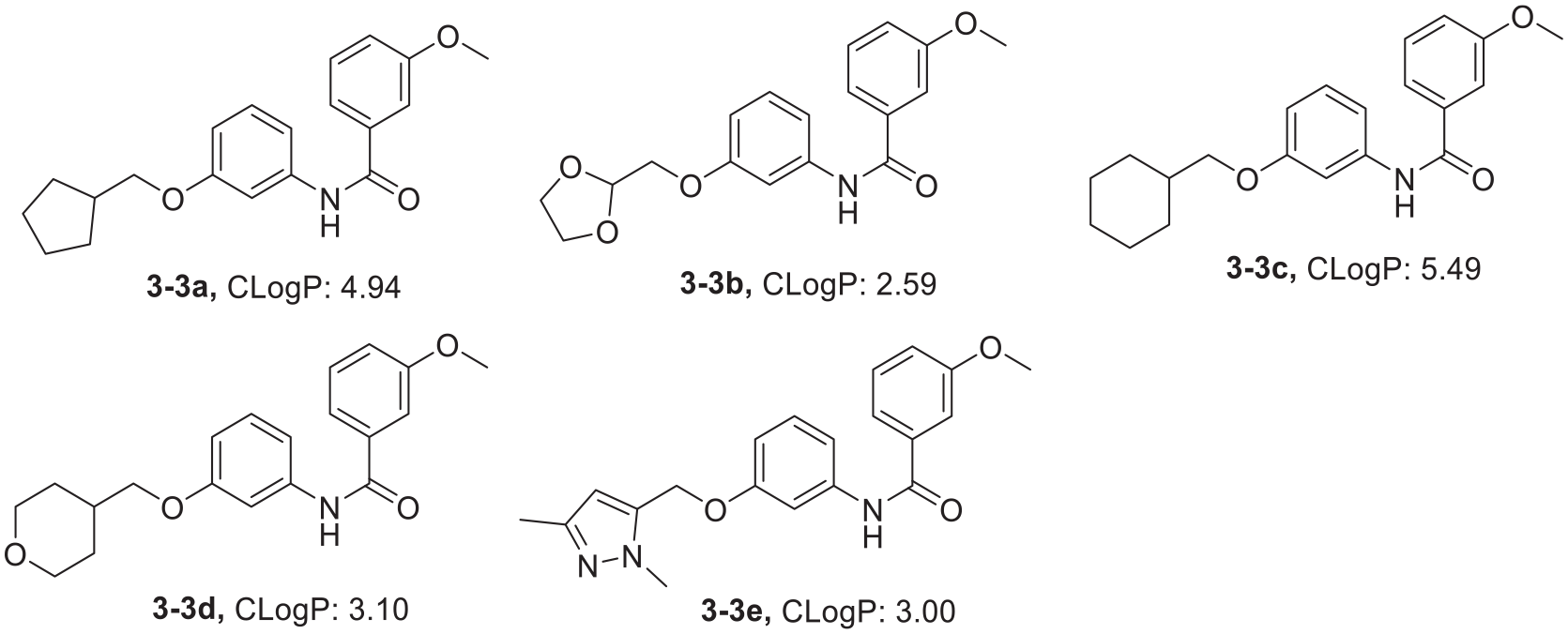
CLogP values of the N-(3-hydroxyphenyl)-3-methoxybenzamide derivatives.
In vitro PDE2 inhibitory activity of the synthesized derivatives.

Bold represents the compound’s higher PDE2 inhibition levels.
BAY60-7550 was used as the reference compound with an IC50 of 8.4 nM.
Results are expressed as the mean of at least three experiments.
Conclusion
Overall, 48 derivatives were designed and synthesized by using UA, UB, and mUA as lead compounds. The inhibitory activity of each derivative against PDE2 was tested at the enzyme level. The results showed that compounds
Methods
Synthesis
All chemicals, reagents, and solvents were of analytical grade and were purchased from commercial suppliers (i.e. Alfa, Meyer, and Aladdin).
1H NMR spectra were recorded on a Bruker DRX spectrometer at 300 and 400 MHz. 13C NMR spectra were recorded on a Bruker BioSpin GmbH spectrometer at 75 MHz, coupling constants are expressed in Hz, MS were recorded on an Agilent 6230 TOFLC/MS mass spectrometer (Agilent, USA). Melting points were measured with an X4-A microscope. Thin-layer chromatography (TLC) was performed on pre-coated silica gel F-254 plates (25 × 75 mm2, Shanghai Pinjia Chemical Co, Ltd.), and samples were observed with ultraviolet light. All raw materials and reagents were purchased from commercial suppliers.
Molecular docking program
DS is a comprehensive scientific prediction platform with the main functions including simulation research, macromolecule design and analysis, antibody development, structure-based design (SBD), fragment-based design (FBD), pharmacophore-based and ligand-based design, QSAR, ADMET, and TOPKAT predicted toxicology, and so on. Its working principle is based on the principles of geometric complementarity, chemical environment complementarity, and energy complementation. If there is an interaction between the ligand and the receptor, the structure–activity relationship and scoring results of the interaction between the receptor and the ligand can be obtained through molecular docking. Taking these results as a reference, it can provide support for the modification of the structure of small molecule compounds. CDOCKER is a flexible docking program based on CHARMM in DS. It can dock ligand molecules with receptor active sites by using soft core potentials and optional grid representation. It is an accurate molecular docking technology, which can produce high-precision docking results. In this paper, the combination of the designed compound with 4HTX was simulated employing the CDOCKER program.
Synthetic methods
Synthesis of 4-hydroxybiphenyl derivatives (1–1a–h )
4-Hydroxybiphenyl (9.4 mmol), K2CO3 (12.2 mmol), R1X (12.2 mmol) and N,N-dimethylformamide (DMF; 30 mL) were added to a flask, and the mixture heated at reflux temperature for 2–24 h. The reaction was monitored by TLC (PE/EtOAc = 5:1). Upon completion, a flocculent was produced after pouring the reaction solution into an ice–water mixture. The crude product was obtained upon filtering and drying. The crude product was dissolved in dichloromethane, and then petroleum ether was added. After standing and filtering, the filter cake was quickly rinsed with dichloromethane. The above operation was repeated two to three times to obtain the corresponding 4-hydroxybiphenyl derivative. 26
5-(([1,1′-Biphenyl]-4-yloxy)methyl)-1-methyl-1H-pyrazole (
5-(([1,1′-Biphenyl]-4-yloxy)methyl)-1,3-dimethyl-1H-pyrazole (
4-[3-([1,1′-Biphenyl]-4-yloxy)propyl]morpholine (
3-(([1,1′-Biphenyl]-4-yloxy)methyl)pyridine (
1-[([1,1′-Biphenyl]-4-yloxy)methyl]-3,5-dimethoxybenzene (
2-(([1,1′-Biphenyl]-4-yloxy)methyl)-1,3-dioxolane (
2-([1,1′-Biphenyl]-4-yloxy) pyrimidine (
2-(2-([1,1′-Biphenyl]-4-yloxy)ethyl)-1,3-dioxane (
Synthesis of 4,4′-dihydroxybiphenyl derivatives 1–2a–f
4,4′-dihydroxybiphenyl (9.4 mmol), K2CO3 (12.2 mmol), R1X (12.2 mmol), and DMF (30 mL) were added to a flask, and the mixture heated at reflux temperature for 2–24 h. The reaction was monitored by TLC (PE/EtOAc = 5:1). Upon completion, a flocculent was produced after pouring the reaction solution into an ice–water mixture. The crude product was obtained upon filtering and drying. The crude product was dissolved in dichloromethane and then petroleum ether was added. After standing and filtering, the filter cake was quickly rinsed with dichloromethane. The above operation was repeated two to three times to obtain the corresponding 4,4′-dihydroxybiphenyl derivative. 26
Ethyl 2-((4′-Hydroxy-[1,1′-biphenyl]-4-yl)oxy)acetate (
4′-(Cyclohexylmethoxy)-4-hydroxy-[1,1′-biphenyl] (
4-Hydroxy-4′-((tetrahydro-2H-pyran-4-yl)methoxy)-[1,1′-biphenyl] (
4-Hydroxy-4′-((tetrahydrofuran-2-yl)methoxy)-[1,1′-biphenyl] (
4-Hydroxy-4′-(3-morpholinopropoxy)-[1,1′-biphenyl] (
4-Hydroxy-4′-(pyrimidine-2-yloxy)-[1,1′-biphenyl] (
Synthesis of 4-hydroxy-4′-methoxybiphenyl derivatives 1–3a–f
4-hydroxy-4′-methoxybiphenyl (9.4 mmol), K2CO3 (12.2 mmol), R1X (12.2 mmol), and DMF (30 mL) were added to a flask, and the mixture heated at reflux temperature for 2–24 h. The reaction was monitored by TLC (PE/EtOAc = 5:1). Upon completion, a flocculent was produced after pouring the reaction solution into an ice–water mixture. The crude product was obtained upon filtering and drying. The crude product was dissolved in dichloromethane and then petroleum ether was added. After standing and filtering, the filter cake was quickly rinsed with dichloromethane. The above operation was repeated two to three times to obtain the corresponding 4-hydroxy-4′-methoxybiphenyl derivative. 26
2-(((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)methyl)tetrahydrofuran (
2-(((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)methyl)-1,3-dioxolane (
4-(((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)methyl)tetrahydro-2H-pyran (
1-(2-((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)ethyl)-1H-pyrazole (
5-(((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)methyl)-1,3-dimethyl-1H-pyrazole (
2-((4′-Methoxy-[1,1′-biphenyl]-4-yl)oxy)pyrimidine (
Synthesis of phenyl 3-methoxybenzoate derivatives 2–1a–f
Step 1: Etherification. Resorcinol (1 mmol), K2CO3 (1.3 mmol), DMF (30 mL), and R1X (0.7 mmol) were added to a flask and the reaction mixture was stirred 80–120 °C for 12 h and monitored by TLC (PE/EtOAc= 4:1). After completion, the reaction solution was poured into an ice–water mixture. The crude product in the aqueous layer was extracted with EtOAc (3 × 100 mL) and the organic layer was filtered and evaporated. The crude product was purified by flash preparative liquid chromatography using petroleum ether and ethyl acetate as the mobile phase. 29 The products, yields, and R1X of the etherification reaction are shown in Table 5.
Step 2: Benzoic acid (1 mmol) was added to dichloromethane(15 mL), the temperature was raised to reflux (40 °C) until the solid had completely dissolved. DMF (1 mL) and SOCl2 (2.2 mmol) were then added dropwise and the mixture stirred at 70 °C for 4 h. The reaction was monitored by TLC (PE/EtOAc = 2:1). After completion, the reaction mixture was directly reduced under vacuum and evaporated to obtain a white solid product. 30
Step 3: Esterification. Tetrahydrofuran (10 mL), the ether from step 1 (1 mmol), and pyridine (300 μL) were added to a flask, and the mixture was stirred at 0 °C for 10 min. Benzoyl chloride (1.1 mmol) was added and the mixture stirred at room temperature for 12 h. The reaction was monitored by TLC (PE/EtOAc = 2:1). After the reaction was complete, suction filtration was performed, and the filtrate was evaporated under reduced pressure. The 3-hydroxyphenyl benzoate derivatives were obtained by using rapid preparative liquid chromatography to purify the product. The mobile phase was petroleum ether and ethyl acetate. 31
The product, yield, and R1X of the unilateral etherification reaction.

3-(Cyclohexylmethoxy)phenyl benzoate (
3-((Tetrahydrofuran-2-yl)methoxy)phenyl benzoate (
3-((Tetrahydro-2H-pyran-4-yl)methoxy)phenyl benzoate (
3-(Pyrimidine-2-yloxy)phenyl benzoate (
3-(2-(1,3-Dioxolan-2-yl)ethoxy)phenyl benzoate (
3-((3,5-Dimethoxybenzyl)oxy)phenyl benzoate (
Synthesis of 3-hydroxyphenyl-3-methoxybenzoate derivatives 2–2a–e
Step 1: See above.
Step 2: 4-Methoxybenzoic acid (1 mmol) was added to dichloromethane(15 mL), the temperature was raised to reflux(40 °C) until the solid had completely dissolved. DMF (1 mL) and SOCl2 (2.2 mmol) were then added dropwise and the mixture stirred at 70 °C for 4 h. The reaction was monitored by TLC (PE/EtOAc = 2:1). After completion, the reaction mixture was directly reduced under vacuum and evaporated to obtain a white solid product. 30
Step 3: Esterification. Tetrahydrofuran (10 mL), the ether from the first step (1 mmol) and pyridine (300 μL) were added to a flask, and the mixture was stirred at 0 °C for 10 min. Next, 3-methoxybenzoyl chloride (1.1 mmol) was added, and the mixture stirred at room temperature for 12 h. The reaction was monitored by TLC (PE/EtOAc = 2:1). After the reaction was complete, suction filtration was performed, and the filtrate was evaporated under reduced pressure. 3-Hydroxyphenyl 3-methoxybenzoate derivatives were obtained by using rapid preparative liquid chromatography to purify the product. The mobile phase was petroleum ether and ethyl acetate. 31
3-((Tetrahydrofuran-2-yl)methoxy)phenyl 3-methoxybenzoate (
3-((1,3-Dioxolan-2-yl)methoxy)phenyl 3-methoxybenzoate (
3-((Tetrahydro-2H-pyran-4-yl)methoxy)phenyl 3-methoxybenzoate (
3-(2-(1H-Pyrazol-1-yl)ethoxy)phenyl 3-methoxybenzoate (
3-((1,3-Dimethyl-1H-pyrazol-5-yl)methoxy)phenyl 3-methoxybenzoate (
Synthesis of N-(3-hydroxyphenyl)benzamide derivatives 3–1a–f
Step 1: Etherification. DMF (20 mL), m-hydroxyaniline (9.2 mmol), NaH (27.5 mmol), and R1X (6.4 mmol) were added to a flask, and the reaction mixture was stirred at room temperature, for 2–18 h. The reaction was monitored by TLC (PE/EtOAc = 4:1). After completion of the reaction, solution was added to 200 mL of water, and then extracted with EtOAc(3 × 100 mL). The solvent was evaporated by rotary evaporation and the residue purified by column chromatography (PE/EtOAc) to give the desired m-hydroxyaniline derivatives. 32 The products, yields, and R1X of the etherification reaction are shown in Table 6.
Step 2: See above. 30
Step 3: Amidation reaction. THF (20 mL), ether from step 1 (12.7 mmol), pyridine (0.2 mL), and benzoyl chloride (16.5 mmol) were, respectively, added to a flask, and the resulting mixture was stirred at room temperature for 14 h. The reaction was monitored by TLC (PE/EtOAc = 3:1). After the reaction was complete, solution was filtered to remove the precipitate, and chromatography (petroleum ether/EtOAc) gave the desired N-(3-hydroxyphenyl)benzamide derivatives. 33
The product, yield, and R1X of the etherification reaction.

N-(3-(2-(1,3-Dioxan-2-yl)ethoxy)phenyl)benzamide (
N-(3-(Cyclohexylmethoxy)phenyl) benzamide (
N-(3-((Tetrahydrofuran-2-yl)methoxy)phenyl)benzamide (
N-(3-((1,3-dioxolan-2-yl)methoxy)phenyl)benzamide (
N-(3-(Pyrimidine-2-yloxy)phenyl)benzamide (
N-(3-((1,3-dimethyl-1H-pyrazol-5-yl)methoxy)phenyl)benzamide (
Synthesis of 3-hydroxy-N-phenylbenzamide derivatives 3–2a–f
Step 1: See above. 32 The products, yields, and R1X of the etherification are shown in Table 6.
Step 2: Preparation of the acid chloride: DCM (50 mL), m-hydroxybenzoic acid (13.8 mmol), and SOCl2 (17.9 mmol) were added to a flask, and the temperature was controlled at 70 °C for 4 h. The reaction was monitored by TLC (PE/EtOAC = 3:1). After the reaction, the solvent was evaporated under reduced pressure to obtain m-hydroxybenzoyl chloride. 30
Step 3: Amidation. The ether from step 1 (12.7 mmol), pyridine (0.2 mL), m-hydroxybenzoyl chloride (16.5 mmol), and THF (20 mL) were, respectively, added to a flask, and the temperature was controlled at room temperature, reacted for 14 h. The reaction was monitored by TLC (PE/ETOAC = 3:1). After the reaction, the reaction solution was filtered to remove the precipitate, and the solvent was evaporated under reduced pressure to obtain a crude product. After column chromatography (PE/ETOAC), 3-hydroxy-N-phenylbenzamide derivatives with higher purity were obtained. 33
N-(3-(Cyclopentylmethoxy)phenyl)-3-hydroxybenzamide (
N-(3-(Cyclohexylmethoxy)phenyl)-3-hydroxybenzamide (
N-(3-(3-dimethoxybenzyl)oxy)phenyl)-3-hydroxybenzamide (
N-(3-((3,5-Difluorobenzyl)oxy)phenyl)-3-hydroxybenzamide (
3-Hydroxy-N-(3-(pyrimidin-2-yloxy)phenyl)benzamide (
N-(3-((1,3-dioxolan-2-yl)methoxy)phenyl)-3-hydroxybenzamide (
Synthesis of N-(3-hydroxyphenyl)-3-methoxybenzamide derivatives 3–3a–e
Step 1: See above.
Step 2: See above. 30
Step 3: Amidation. THF (20 mL), the etherification product (12.7 mmol) from step 1, pyridine (0.2 mL) and 4-Methoxybenzoic chloride (16.5 mmol) were, respectively, added to a flask, and the resulting mixture was stirred at room temperature for 14 h. The reaction was monitored by TLC (PE/EtOAc = 3:1). After the reaction was complete, solution was filtered to remove the precipitate, chromatography (petroleum ether/EtOAc), the desired N-(3-hydroxyphenyl)-3-methoxybenzamide derivatives were obtained. 33
N-(3-(Cyclopentylmethoxy)phenyl)-3-methoxybenzamide (
N-(3-((1,3-dioxolan-2-yl)methoxy)phenyl)-3-methoxybenzamide (
N-(3-(Cyclohexylmethoxy)phenyl)-3-methoxybenzamide (
3-Methoxy-N-(3-((tetrahydro-2H-pyran-4-yl)methoxy)phenyl)benzamide (
N-(3-(1-dimethyl-1H-pyrazole-5-yl)methoxy)phenyl)-3-methoxybenzamide (
PDE2 enzyme inhibitory activity experiment
BAY60-7550 was employed as a positive control, phosphodiesterase II (PDE2) was used as the target, and the compounds
Supplemental Material
sj-docx-1-chl-10.1177_17475198221148080 – Supplemental material for Design, synthesis, and biological evaluation of urolithin derivatives as potential phosphodiesterase II inhibitors
Supplemental material, sj-docx-1-chl-10.1177_17475198221148080 for Design, synthesis, and biological evaluation of urolithin derivatives as potential phosphodiesterase II inhibitors by Hecheng Wang, Long Tang, Wanhui Di, Feng Yan, Xianfeng Huang, Xiaoqing Feng and Guoqiang Song in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.