
Abstract
To improve the successful prediction rate of the existing molecular docking methods, a new docking approach is proposed that consists of three steps: generating an ensemble of docked poses with a conventional docking method, performing clustering analysis of the ensemble to select the representative poses, and optimizing the representative structures with a low-cost quantum mechanics method. Three quantum mechanics methods, self-consistent charge density-functional tight-binding, ONIOM(DFT:PM6), and ONIOM(SCC-DFTB:PM6), are tested on 18 ligand-receptor bio-complexes. The rate of successful binding pose predictions by the proposed self-consistent charge density-functional tight-binding docking method is the highest, at 67%. The self-consistent charge density-functional tight-binding docking method should be useful for the structure-based drug design.

Introduction
A significant task of rational drug design is to determine the three-dimensional (3D) structure of a drug candidate that binds to a particular bio-macro-molecule. Due to the complexity of the bio-complex potential energy landscape involving a gigantic number of local minima,1,2 the binding mode structure is often determined by molecular docking simulations. 3 A docking algorithm generates possible ligand conformations within the receptor-binding site by some conformational sampling technique,1,4 and a quick-to-evaluate empirical scoring function estimates the corresponding binding free energy energies. 5 However, while the molecular docking technique can quickly generate and evaluate a huge number of docked pose conformations, the obtained binding pose structure is often not very accurate. 6
The inaccurate docking results are caused mainly by the inadequate scoring function for the estimations of the binding free energies. To overcome the shortcoming of the empirical scoring function, Rao et al. 7 proposed a so-called DOX method to predict non-X-ray protein-ligand binding structures. The docking-generated poses are first screened by a scoring function in the DOX method. The chosen poses are then ranked by the single-energy calculations of ONIOM3 (ωB97X-D 8 /6-31G*:PM6: 9 Am-Amber).10,11 The top 10 ONIOM3-ranked poses are further optimized with the XO2 (ωB97X-D/6-31G*:PM6)12,13 calculations. The optimal binding structure is finally determined at the XO2 (ωB97X-D/6-311+G**:PM6) level of theory. Tests show that the DOX method is capable of providing very good results. The DOX method is recently improved and refined as a CSAMP strategy that provides excellent binding structures. 14 Despite the impressive results of the DOX-CSAMP method, the method is computationally demanding, which is a serious obstacle for its wide applications.
A compromise between accuracy and efficiency is required for the practical use of a computational method. The CPU cost of the DOX protocol is rooted in the density-functional tight (DFT) calculations of the ONIOM method. Minimizing the cost of DFT computations is key to the numerical efficiency of predicting the ligand binding structure. The density-functional tight-binding DFTB theory15,16 is a second-order approximation of DFT. DFTB is 100–1000 times faster than DFT but is widely tested to produce results similar to DFT.17,18 Therefore, the effort of exploring the use of DFTB for the binding pose prediction of bio-complex is significant.
This work aims to test the quality of using self-consistent charge DFTB (SCC-DFTB) to predict ligand binding structures. The performances of molecular docking, SCC-DFTB geometry optimization, ONIOM(DFT:PM6), and ONIOM(SCC-DFTB:PM6) are compared for 18 crystal protein-ligand binding poses. The SCC-DFTB predictions are surprisingly good when compared with the experimental results.
Computational methods
Prediction of the binding poses of protein-ligand complexes
The workflow of the binding pose prediction consists of three steps (Figure 1): (1) generation of an ensemble of ligand conformations by a conventional docking method (classical docking), (2) clustering analysis of the ensemble to select the representative poses, and (3) optimization of the representative structures by a quantum mechanics (QM)-based method to determine the binding poses. The technical details of the procedure are given as follows.

Workflow of the proposed quantum mechanics (QM)-based docking method. The QM method used here refers to one of the following: SCC-DFTB, ONIOM(SCC-DFTB:PM6), and ONIOM(ωB97X-D/6-31G*:PM6).
Classical docking
The binding pose of a given protein-ligand complex is first predicted by performing the widely used molecular rigid docking simulations, referred as the classical docking. The classical docking is carried out with the MOE-Dock software 19 with its default settings. That is, the ligand conformations are generated by the bond rotation method (the setting of “Rotate Bonds”), using the setting of “Triangle Matcher” for the placement method and “London dG” for scoring. Then, the complex structures are improved by the refinement method of “Rigid Receptor” and scored with the parameter setting “Forcefield/GBVI-WSA dG.” The retained values of both the placement and refinement methods are 200.
At the end of classical docking, the docked poses are saved into the MOE database and sorted/ranked according to the scoring function in MOE. The ensemble of classical docking poses is used as the starting point of quantum docking, a term used here to refer broadly to a method of binding pose prediction involving some QM calculations.
Following the classical docking, the quantum docking includes two main steps: (1) ensemble clustering analysis and (2) binding pose prediction by QM calculations.
Ensemble clustering analysis
To minimize the computational cost of quantum docking, the set of classical docking poses is first grouped into clusters of similar structures by an ensemble clustering analysis. The clustering analysis is conducted through the UCSF Chimera program 20 applying the procedure of Kelley et al. 21 The program uses the pair-wise best-fit root mean square deviation (RMSD) to select a conformation to represent a cluster with more than one member. Only the cluster representing conformations are considered for QM calculations. Notice that a cluster is ignored here if it contains only one member.
Binding pose prediction by QM calculations
The cluster representing poses is merged with the receptor’s active binding site analog to form candidate binding modes. The receptor analog is a reduced version of the receptor, used to faithfully reflect the ligand-receptor interaction while reducing the cost of QM computation. It is obtained by considering the amino acid (AA) residues of the receptor within 6 Å from the ligand. There are about 700 atoms in a receptor analog.
The ligand geometries of the candidate binding modes are re-docked (geometry optimization) by QM calculations. Three types of QM calculations, SCC-DFTB, ONIOM(DFT:PM6), and ONIOM(SCC-DFTB:PM6), are performed, referred as the SCC-DFTB docking, the DFT:PM6 docking, and the SCC-DFTB:PM6 docking, respectively. Alternatively, they are for simplicity referred to as the QML docking, the QMH:PM6 docking, and the QML:PM6 docking, respectively. The SCC-DFTB method is applied to the whole ligand-receptor analog system in the QML docking. For example, in the QMH:PM6 docking, the ligand is treated at the level of ωB97X-D/6-31G*, while the receptor analog is treated with the PM6 method. Similarly, the SCC-DFTB theory treats the ligand, and the receptor analog is treated by the PM6 method in the QML:PM6 docking.
The re-docked binding modes are ranked simply by their final single-point energies, that is, the entropy effect is assumed to play a minor role and ignored to save the computational cost. The docked mode with the lowest energy is chosen as the binding pose of the protein-ligand complex.
The QM calculations are carried out through the Gaussian09 package 22 using the loose criterion for convergence, which permits the use of the full functionalities of SCC-DFTB. The SCC-DFTB calculations use the parameter sets of 3OB 23 and MIO 24 for the first-row and transition elements.
The qualities of the classical and quantum docking results are assessed by the RMSDs between the predicted poses and the crystal pose. Only the heavy atoms of the ligands are considered in the RMSD computations. The RMSDs are calculated using the Kabsch algorithm. 25 Following the common practice, the possibility of flexible groups of ligands affecting the RMSD computations is ignored as it is considered unlikely due to the receptor-ligand interactions. Python script of this algorithm (https://github.com/charnley/rmsd) is utilized to detect the correct pairing between the same type of atoms. A docking result has been deemed a success if the calculated RMSD is smaller than 2 Å,26,27 although some other studies may use the criterion of RMSD less than 2.5 Å. 28
Preparation of protein-ligand data sets
18 X-ray crystal structures of receptor-ligand complexes in the PDB format are downloaded from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). 29 The receptor PDB IDs are 3NHX, 30 1FKI, 31 1ERB, 32 2QI3, 33 2I0A, 34 2ANL, 35 1A4K, 36 2BJU, 37 1HWK, 38 1HWJ, 38 3CCW, 39 3CD0, 39 1FKG, 31 3MXD, 40 3CD7, 39 1FKH, 31 3GI5, 41 and 3BGL. 42 The corresponding ligand PDB IDs are ASD, SB1, ETR, MZ5, MUI, JE2, FRA, IH4, 117, 116, 4HI, 6HI, SB3, K53, 882, SBX, K62, and RID, respectively. General information about the protein-ligand pairs can be found in Table S1 in the Supporting Information. Basic properties such as the number of rotatable bonds, topological polar surface area, and molecular weight of the 18 ligands are shown in Table S2 in the Supporting Information. Non-covalent bonds bind all the 18 receptor-ligand complexes. Besides, their DFTB parameters are fully available from www.dftb.org. 43
When preparing the bio-complexes for molecular docking, water molecules and ions present in the crystal structures are detached. Missing atoms or residues that should exist in the crystallographic structure are added to their structures using MOE 2015.10. 19 Each bio-complex is assigned at the physiological conditions for titratable groups using the Protonate 3D module in MOE. The bio-complexes are then subjected to energy minimization (EM) using amber01: 44 EHT 45 molecular mechanics force field with the default settings of R-Field solvation model, Rigid Water Molecules Constraint, and 0.1 kcal/mol/A2 for the gradient RMS cut-off.
Following EM, the binding site of the receptor AAs is identified using the Surface and Maps operations in MOE, with the setting “Molecular Surface” within 6 Å of the ligand atoms.
Results and discussion
Ensemble cluster analysis
The automated classical docking simulations with the MOE-Dock program generate 200 docked poses for each bio-complex. The MOE-Dock program is also used to remove the possible duplication of docked poses. The removing operation is required as the portion of duplicated docking poses can be quite high for inflexible ligands. For example, only 38 and 113 unique poses are left respectively for the two ligands of SB1 and ASD that have no rotatable bonds (Table S2). However, the number of duplicated docking poses removed for most of the 18 ligands is quite small, as can be inferred from Figure 2.

Number of the docked pose clusters and the ratio (percentage) of the number of poses in the clusters (as shown in parenthesis) to the number of all unique poses for each of the 18 ligands.
The sets of remaining unique docked poses for the ligands are then subjected to the ensemble clustering analysis. A substantial amount of one-pose clusters are found. The one-pose clusters are discarded after the clustering analysis as the probability of finding the best pose in them is low. For convenience, unless explicitly stated otherwise, the term “cluster” is used below to refer a cluster with at least two docked poses.
The results of the clustering analysis of the docked pose ensembles of the 18 ligands are summarized in Figure 2. As shown in Figure 2, except for ASD and SB1, the obtained clusters consist of over 90% of the docked poses for anyone of the ligands. The special results for ASD and SB1 are easy to understand. As mentioned above, there are no rotatable bonds in ASD and SB1. The inflexibility of ASD and SB1 leads to many of their docked poses belonging to the one-member cluster that are ignored in this study. As a result, the docked pose coverages of the clusters of ASD and SB1 are relatively low, but still higher than 70%. Overall, the clusters are highly representative of the docked pose ensemble.
The number of poses in a cluster can be very different for different clusters. For example, the number of poses in a cluster varies from 3 to 31 for MUI. A cluster with a large number of poses indicates that the structural feature shared by the cluster poses is assessed favorable by the scoring function. A cluster with more poses should be more important than a cluster with fewer poses, if the scoring function is perfect. Unfortunately, the scoring function in MOE is far from ideal and it is hard to prejudge the importance of a cluster. Consequently, all clusters are treated equally. One pose, the central pose of a cluster, for each cluster is used for further QM geometry optimization and ranking.
Docking results and performance comparison
The RMDSs between the crystal binding poses and the top docked poses predicted by different docking methods are shown in Table 1.
RMSDs (in Å) of the top docked pose structures as predicted by the classical docking, SCC-DFTB (QML), ONIOM(QMH:PM6), and ONIOM(QML:PM6) docking methods.
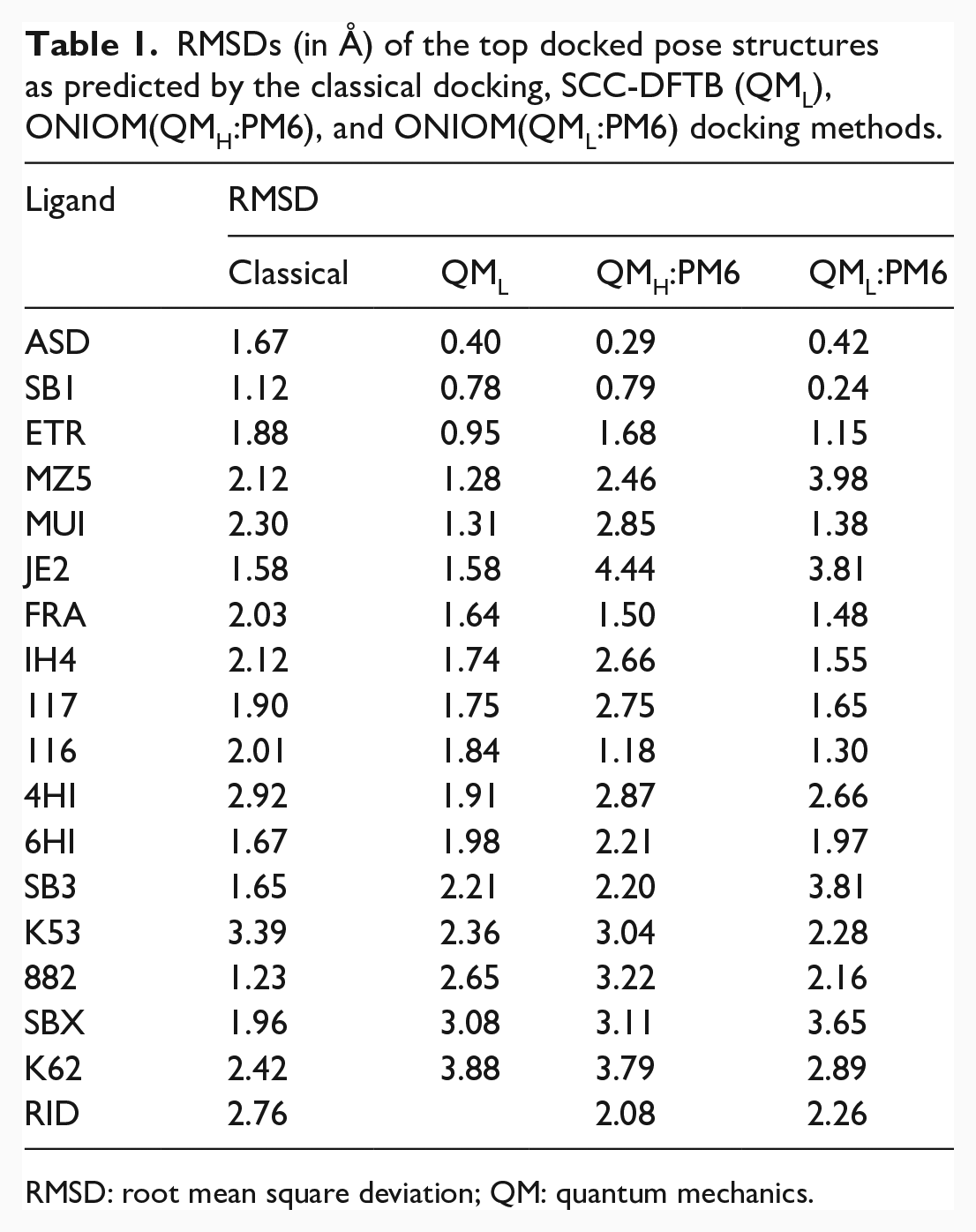
RMSD: root mean square deviation; QM: quantum mechanics.
As seen in Table 1, 9 out of the 18 top-scored classical docking poses have RMSD less than 2 Å. The classical docking method has a success rate of 50%. The largest and smallest RMSDs of the classical docking method are 3.39 Å for K53 and 1.12 Å for SB1, respectively. The average RMSD for the 18 ligands is 2.04 Å. The average RMSD for the 9 successfully predicted ligands is 1.63 Å.
The number of ligands with RMSD less than 2 Å is increased to 12, or a success rate of 67% for the QML (SCC-DFTB) docking method. Not only has the SCC-DFTB docking increased the rate of successful docking, but it has also improved the quality of docking. The average RMSD for the 12 SCC-DFTB successfully docked systems is 1.43 Å, compared with 1.63 Å for the nine systems of successful classical docking.
The missing entry in Table 1 concerning the QML docking of RID is caused by the non-convergent geometry optimization cycle of the SCC-DFTB calculations. The SCC-DFTB calculations on the candidate docked poses as deduced from the ensemble cluster analysis were unsuccessful for the three cases of K53, K62, and RID. As a remedy, the candidate binding structures were further relaxed using the PM3 method46,47 and then used as the starting geometries for the SCC-DFTB docking. The SCC-DFTB docking results thus obtained for K53, K62, and RID are shown in Table 1. However, as seen in Table 1, although the SCC-DFTB calculations for K53 and K62 with the modified starting structures run properly, the obtained docking results, with the RMSDs of 2.36 and 3.88 Å, are poor. That is, the attempted remedy is unsuccessful and unnecessary in practical applications.
According to the data shown in Table 1, the number of ligands with RMSD less than 2 Å is only five, or a success rate of 28% for the QMH:PM6 docking method. The success rate of the QML:PM6 docking is 50% or the same as the classical docking. Overall, the qualities of the docking methods are ordered as SCC-DFTB > QML:PM6 ≈ classical > QMH:PM6. The SCC-DFTB docking produces the best results, while the QMH:PM6 docking yields the worst results. Concerning the computational time, t, t(QMH:PM6) ≈ 1.5 t(SCC-DFTB) ≈ 6 t(QML:PM6). The SCC-DFTB docking is four times slower than the QML:PM6 docking but is one-third faster than the QMH:PM6 docking.
The SCC-DFTB docking results with a success rate of 67% is highly satisfactory. However, the poor performance of the QMH:PM6 docking with a success rate of only 28% is quite unexpected. For the purpose of understanding the underlying mechanism, Figure 3 shows a comparison of the RMSDs of the starting and final docked poses of the 18 bio-complexes for the three QM docking methods. A starting pose refers to the starting geometry from which a predicted final top pose is generated and is dependent on the specific docking method. As shown in Figure 3, the SCC-DFTB geometry optimization usually results in final poses with lower RMSDs compared to the starting poses, specifically for 12 of the 18 cases. However, the ONIOM(DFT:PM6) geometry optimization increases the final pose RMSD for 11 of 18 cases, yielding the poor results of the QMH:PM6 docking method seen above. The ONIOM(SCC-DFTB:PM6) optimization increases (decreases) the final pose RMSD for 10 (8) of 18 cases, correlating with the observed medium-quality results of the QML:PM6 docking.

Comparisons of RMSDs of the starting and final poses of three QM docking methods.
The geometry optimization results indicate that it is preferable to use a computational method that treats the receptor-binding pocket and the ligand at a similar level of theory. The SCC-DFTB docking that treats the binding pocket and the ligand at the same level of theory yields the best results. The two-layer ONIOM calculations treat a small region of the system, only the ligand, at a higher level of theory and the rest at a lower level of theory. The difference between the electronic descriptions of SCC-DFTB and PM6 is smaller than that between DFT and PM6. Therefore, the combination of SCC-DFTB and PM6 in the ONIOM geometry optimization yields better results than that with the DFT-PM6 combination. Notice that the above observations are fully consistent with the findings of Lundberg et al. 48 who concluded that (1) DFTB is capable of describing the interaction between subsystems well and (2) the ONIOM approach using very different levels of theory may produce poor results.
Although the SCC-DFTB method produces the best overall results among all the methods examined, its results for some ligands are inferior to the classical docking, as may been in Table 1. The finding that some QM results are worse than the classical ones is in fact not surprising. The proposed method is required to have an affordable computational cost for practical applications. For that reason, the proposed method involves numerous approximations that are absent in classical docking. The major approximations include (1) all one-member clusters are neglected to save the cost of QM computation, which may sometime reduce the coverage of ligand conformations substantially. (2) Only the central pose of a cluster is chosen for further QM study. The central poses are not always the best representations of their clusters and do not lead to the possible topmost poses. (3) The entropic effect is assumed to be inconsequential for the ranking of different poses. The assumption may lead to erroneous predictions when very different ligand conformations are involved. 49 (4) The solvation effect is not considered in the QM calculations. The solvation effect is known to significantly reduce the energy differences among different binding modes, but often maintains their relative energy ordering. 50 However, the conformational energy ordering can also be easily changed if the ligand-solvent interactions are quite different for different ligand poses. 51 Despite all the imperfections involved, it is comforting to see that the proposed method possesses the merit of providing a high rate of successful binding pose prediction with a modest level of computational cost.
Summary
Increasing the success rate of ligand-receptor docking is proposed and tested on 18 ligand-receptor bio-complexes. Starting from the representative poses of conventional docking simulations, the geometry optimizations with the SCC-DFTB method yield a docking success rate of 67%. Considering the high success rate and reasonable computational cost, the proposed SCC-DFTB docking method can be useful for structure-based drug design.
Supplemental Material
sj-doc-1-chl-10.1177_17475198221101999 – Supplemental material for Combining classical molecular docking with self-consistent charge density-functional tight-binding computations for the efficient and quality prediction of ligand binding structure
Supplemental material, sj-doc-1-chl-10.1177_17475198221101999 for Combining classical molecular docking with self-consistent charge density-functional tight-binding computations for the efficient and quality prediction of ligand binding structure by Amar Y Al-Ansi, Haorui Lu and Zijing Lin in Journal of Chemical Research
Footnotes
Acknowledgements
The authors acknowledge the computing time of the Supercomputing Center of the University of Science and Technology of China.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (12074362 and 11774324).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.