
Abstract
Trans unsaturated fatty acids in humans may be originated both from dietary supplementation and from an endogenous free-radical-catalyzed cis−trans isomerization of fatty acid residues in naturally occurring cis lipids. The latter process affords geometrical isomers and the polyunsaturated fatty acid mono-trans isomers were demonstrated to be connected with stress conditions in living organisms. Synthesis of mono-trans polyunsaturated fatty acid is useful for analytical and biological research, and in this case, the availability of free fatty acids is needed as well as the possibility of mg scale of the synthetic protocol. Herein, we report a simple synthetic route to mono-trans isomers of arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid, which includes thiyl radical-catalyzed isomerization reaction of polyunsaturated fatty acid methyl esters and fraction isolation of mono-trans mixture isomers followed by optimization of hydrolysis condition to free fatty acids and purification of each mono-trans polyunsaturated fatty acid. Our approach to mono-trans polyunsaturated fatty acids as free acids can reach the mg scale, thus fostering more applications to biochemical and biological studies.
Introduction
Trans fatty acids (TFAs) are by-products of industrial processes, such as oil deodorization or partial hydrogenation which are frequently used to improve the palatability of dietary ingredients. 1 Investigation of TFAs in the last two decades in our laboratory established the occurrence of thiyl radical (RS•)-catalyzed isomerization of the natural cis lipids into the corresponding geometrical trans isomers.2,3 Thiyl radicals that can be formed also in vivo during cellular stress can explain the endogenous origin of trans unsaturated fatty acids in various contexts: cells,4,5 animals,6,7 and humans.8–10 In these cases, mono-trans polyunsaturated fatty acid (mt-PUFA) isomers were evidenced as the most probable products of free radical attack in biological conditions, thus pointing their significance as biomarkers, as shown, for example, for arachidonic acid isomers.6,9,11 TFAs are responsible for multiple cell dysfunctions and disease outcomes, mostly due to interferences of biological processes given by the change of the natural cis geometry of unsaturated lipids to the unnatural trans one.12,13 The importance of TFA detection motivated research in synthetic approaches and analytical protocols to allow their precise separation, detection, and quantitation. The thiyl radical-based synthetic approach, used by our group for the preparation of several geometrical TFA of monounsaturated fatty acid (MUFA) and PUFA, occurs via the addition of thiyl radical to one double bond, rotation of C–C bond in the adduct radical, and β-fragmentation of thiyl radical, as shown in Scheme 1.14–16 In PUFA, the time profiles for the disappearance of linoleic acid (9c,12c−C18:2) and arachidonic acid (5c,8c,11c,14c-C20:4) methyl esters and formation of mt-PUFA isomers gave the first indication that the isomerization occurs stepwise.11,17 Such reactivity has been recently investigated in complex lipids like cardiolipins, giving again evidence of the formation of mt-cardiolipins as first isomers. 18

The formation of mt-PUFA can be obtained under acidic conditions by para-toluensulfinic acid as performed for arachidonic acid (ARA) 19 and eicosapentaenoic acid (EPA). 20 In the former case, the ARA isomers were used to characterize the •NO2 radical-mediated isomerization that was induced in vivo. Other methods implied the use of metabolic transformation in the cellular or algal environment, starting from the trans precursors (linoleic and alpha-linolenic acids) which are commercially available21–23 or the use of high temperature in PUFA-containing oils. 24 The first regio- and stereo-specific synthesis of mt-PUFA isomers was reported for ARA via the mono-epoxidation and purification of the four mono-epoxide isomers and subsequent elimination of the epoxide functionality with the formation of the trans double bond. 25 This strategy was used subsequently by us to form mt isomers of the natural omega-3 eicosapentaenoic acid (5c,8c,11c,14c,17c-C20:5, EPA) and docosahexaenoic acid (4c,7c,10c,13c,16c,19c-C22:6, DHA)26,27 but in the form of methyl esters. In fact, the pure mt isomers of free fatty acids were not obtained, and we understood that optimization of hydrolysis conditions is still an important issue for PUFA. In fact, for mt-ARA as free acid, we previously reported the direct isomerization of ARA acid by thiyl radical-catalyzed isomerization, 28 followed by purification via silver-complexation. 29 In this procedure, the conversion of mt-ARA to methyl ester, to perform the purity assessment by gas chromatography (GC), was performed by a mild, though not easy procedure, by an explosive reagent such as diazomethane, to convert free acids into methyl esters. Indeed, the fatty acid isomer identification is mainly performed by GC and GC/MS, able to separate geometrical and positional isomers, in the form of fatty acid derivatives, such as methyl esters (FAME). We published optimal detections of ARA, EPA, and DHA mt-isomers as methyl esters by this methodology,17,26,27 and also others have described successful separation in this case. 30 By GC, free fatty acids cannot be eluted; therefore, the use of other analytical tools is required, and recently, some efforts have been made in the area of trans isomer separation.
High-performance liquid chromatography (HPLC) with stationary reverse phase columns can be useful for lipid separation and was recently found to distinguish cis and trans isomers when coupled with online ozonolysis and ion mobility-mass spectrometry. 31 It is worth noting at this point that the identification of geometrical isomers cannot be confused with the identification of positional isomers, where the change of the double bond position, not the geometry, is involved. Indeed, such positional isomers were recently separated using LC-MS by solvent plasmatization and the interaction with the ion source, which facilitate epoxidation or peroxidation of carbon-carbon double bond, thus providing for characteristic molecular fragmentations via collision-induced dissociation (CID), elucidating the position of the double bond. 32 Another liquid chromatography condition that separates cis and trans fatty acid isomers is Ag+-HPLC, largely used for MUFA in alimentary samples. 33 Further chromatographic methodologies were reviewed for the separation of positional and geometrical fatty acid isomers based on the application of TLC, GC, HPLC, and supercritical fluid chromatography (SFC). 34
In view of ameliorating the availability of mt-PUFA as free acids, we present herein a revised protocol for an mg scale synthesis of mt-PUFAs of ARA, EPA, and DHA as shown (Figure 1) that facilitates biochemical and biological studies and provides references for analytical purposes.

Chemical structures of ARA, EPA, and DHA; in red are the positions of carbon-carbon double bonds. On the right side, the total number of possible mt isomers is reported.
Results and discussion
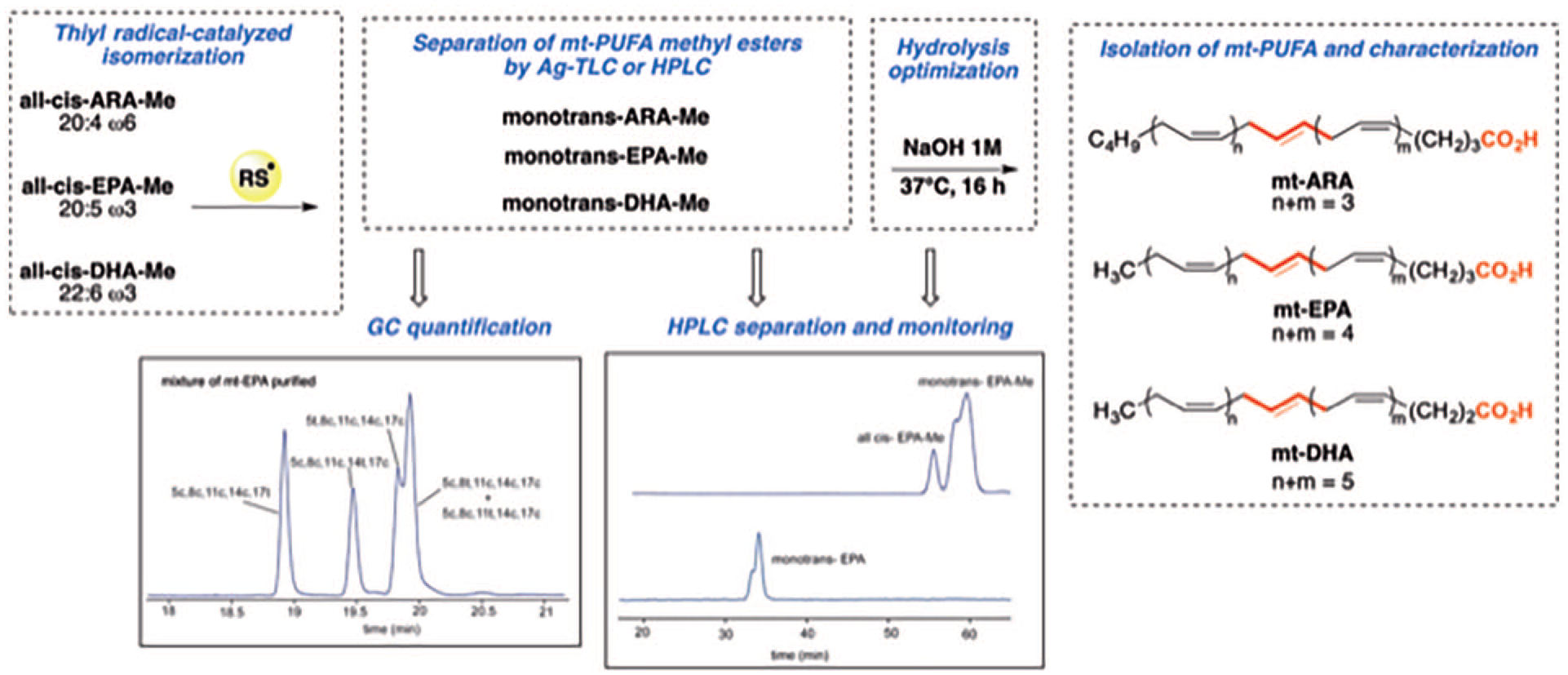
We started from the corresponding commercially available methyl esters (ARA-Me, EPA-Me, and DHA-Me) that have been successfully studied by us for the formation of the corresponding mt-PUFA-Me.8,9,26,27 In Figure 2, the steps of our revised approach to mt- PUFA are shown, as follows: (1) preparation of mt-PUFA-Me by thiyl radical catalyzed cis-trans isomerization, (2) isomeric fraction isolation by argentation method or semi-preparative reverse phase HPLC, and (3) optimization of hydrolysis procedure and purification of mt-PUFA.

Flow chart for the preparation of mt isomers of PUFA as free fatty acids (mt-PUFA).
Synthesis of mt-PUFA-Me
The cis-trans isomerization of ARA-Me, EPA-Me, and DHA-Me (15 mM in i-PrOH) was performed by photolysis at λ = 250–260 nm in the presence of β-mercaptoethanol (7.5 mM) in a micro photochemical reactor, following our previously reported methodology for such transformation 18 and as described in section “Materials and methods.” The crucial point of this protocol was to keep the reaction time long enough to form an appreciable amount of mt isomers, but at the same time to avoid the formation of di-trans and poly-trans isomers. Their formation would indeed complicate the following purification step. By performing several control experiments we set the UV-irradiation time to 5 min to achieve an overall conversion yield of the total mt isomers of ca. 30%. Quantification was performed through the FAME transformation of an aliquot of the reaction mixture and analysis under known GC conditions. GC retention times (RTs) are reported in Table 1 (see section “Materials and methods” for GC conditions). Mt isomers were formed in different amounts for each PUFA, depending on the position of the double bonds.
Geometrical mt isomers of ARA-Me, EPA-Me, and DHA-Me and corresponding retention times in GC analysis (conditions described in section “Materials and methods”). See Supplementary Information for the GC runs (Figures S1–S3 in Supplementary Information).

ARA-Me: arachidonic acid methyl ester; DHA-Me: docosahexaenoic acid methyl ester; EPA-Me: eicosapentaenoic acid methyl ester; mt: mono-trans.
RT: retention time in minutes.
Not completely resolved.
The reaction mixture was then used to separate mt isomers from the unreacted all-cis PUFA-Me. We used the preparative Ag-TLC under appropriate elution conditions to isolate the desired mt mixtures of ARA-Me. The purity of the obtained products was determined by GC analysis (>98.4%) and the four mt isomers were present in approximately equal amounts as expected in solution.11,17 We also explored the efficiency of semipreparative RP-HPLC, using a C18 column, under optimal elution conditions to guarantee the separation of the peaks of ARA-Me, EPA-Me, and DHA-Me cis–trans and mt mixtures. In this system, we found that RTs of the FAME were distinguishable from the corresponding mt-PUFA (Table 2, entries 1 and 2) (see Figures S4–S6, Supporting Materials). We obtained pure mt-PUFA fractions for ARA-Me, EPA-Me, and DHA-Me via semipreparative HPLC multiple runs (10 µL injection volume) by using a fraction collector based on the UV signals (threshold 7 mAU). Purity for mt-EPA-Me and mt-DHA-Me was very good (>99% and >98.4%, respectively) as investigated by the transformation of each fraction in FAME and GC analysis.
Reverse-phase HPLC separation of PUFA-Me (all-cis) and mt-PUFA-Me and the corresponding mt-PUFA. See Supplementary Information for HPLC runs (cf. Figures S4–S6 in Supplementary Information).

ARA: arachidonic acid; DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid; HPLC: high-performance liquid chromatography; Me: methyl ester; Mt: mono-trans; PUFA: polyunsaturated fatty acid.
RT: retention time in minutes.
Broad peak.
Multipeaks not resolved.
Synthesis of mt-PUFA as free fatty acids
Having the mt fatty acids fraction in the form of Me, the hydrolysis step is needed to finally obtain the mt-PUFA of interest.
Over the years, many synthetic procedures were reported for the hydrolysis of fatty acids, using either acidic or basic reaction conditions. Taking into account the reactivity of polyunsaturated substrates like PUFAs, we decided to perform a detailed experimental optimization to reduce any possible side-reaction that influences the yield. Such optimization was carried out using the ARA-Me and the results are shown in Table 3 using two different (acidic and alkaline) conditions.
Optimization of the hydrolysis conditions using 1 mg mL−1 of ARA-Me.
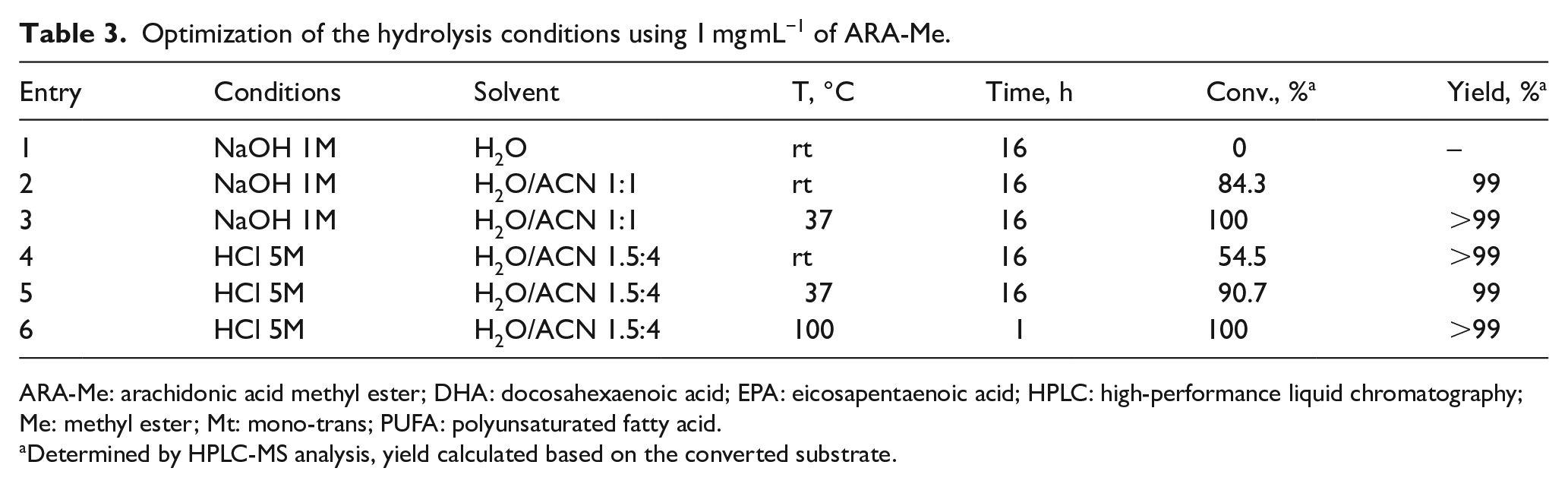
ARA-Me: arachidonic acid methyl ester; DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid; HPLC: high-performance liquid chromatography; Me: methyl ester; Mt: mono-trans; PUFA: polyunsaturated fatty acid.
Determined by HPLC-MS analysis, yield calculated based on the converted substrate.
By using 1 M NaOH aqueous solution, hydrolysis proceeded smoothly at room temperature for 16 h employing ACN as co-solvent (1:1 with the aqueous system of the alkaline reagent), without any formation of by-products with a conversion yield >84%. Complete conversion of the starting material was obtained by an increase in the reaction temperature to 37 °C for 16 h. HPLC analysis showed a complete conversion and a yield of >99% (Table 3, entry 3). Conversely, we used acidic conditions created by 5 M HCl aqueous solution, according to a procedure reported in 2017 by Takashima et al. for extraction of fatty acids by hydrolysis in biological samples. 35 One milligram of ARA-Me was dissolved in 0.73 mL of ACN and 0.28 mL of 5 M HCl in water. The reaction mixture that was stirred at room temperature for 16 h had a conversion yield of 54.5% (entry 4, Table 3), whereas at 37 °C for 16 h, a conversion yield of 91% was obtained (Table 3, entry 5). To shorten the reaction yield, a temperature of 100 °C was used for 1 h, finding a complete conversion, >99% yield (Table 3, entry 6).
We selected the mild temperature of 37 °C and the alkaline condition as optimized hydrolysis of the mt-PUFA-Me mixtures. The protocol was successfully applied to mt-ARA-Me, mt-EPA-Me, and mt-DHA-Me, isolating the target mixtures of mt free fatty acids with the same excellent purity. The RT at HPLC analysis is shown in Table 2, and MS spectra confirmed the purity of the mt-PUFA as free fatty acids (see Figure S3–S7 in Supporting Materials).
Conclusion
The synthesis of mt fatty acids was carried out for some relevant PUFA: ARA, EPA, and DHA for their involvement in important biological processes. The procedure can be performed with standard equipment of organic synthesis and leads to the mg scale of these isomers useful for further use both as a standard reference in analytical protocols and as substrates for enzymatic and metabolic studies needed to deepen the understanding of the role of lipid geometry and the consequences of isomeric transformation in living organisms.
Material and methods
PUFA-Me were analyzed by GC (Agilent 6850, Milan), using the split mode (50:1), equipped with a 60 m × 0.25 mm × 0.25 µm Zebron column (Phenomenex, Torrence, CA, USA), and a flame ionization detector with the following oven program; temperature started from 165 °C, held for 3 min, followed by an increase of 1 °C min−1 up to 195 °C, held for 40 min, followed by a second increase of 10 °C min−1 up to 240 °C, and held for 10 min. A constant pressure mode (29 psi) was chosen with hydrogen as the carrier gas. The mt-PUFA-Me was identified by comparison with authentic samples and chromatograms were examined as described previously.8,9,26,27 Figures S1–S3 in Supplementary Materials show the mt-ARA-Me, mt-EPA-EPA, and mt-DHA-Me in the GC runs.
UV irradiations were performed in a micro photochemical reaction assembly with quartz well (Ace Glass) using a 5.5 W cold cathode, low-pressure, mercury arc, gaseous discharge lamp (corresponds to λ = 250–260 nm) made of double-bore quartz (Ace Glass).
HPLC-MS analysis were performed with an Agilent 1260 Infinity II HPLC system equipped with a 5-µm C18 column (25 × 4.6 mm) using as eluent: MeOH/H2O+0.2% formic acid 8:2, 1 mL min−1, injection 5 µL, coupled with an InfinityLab single quadrupole Liquid Chromatography/Mass Selective Detector (LC/MSD) and with a diode array detector set at 204 nm. LOQ (ARA) = 0.0013 mg mL−1, LOD (ARA) = 0.004 mg mL−1, LOQ (ARA-Me) = 0.0013 mg mL−1, LOD (ARA-Me) = 0.004 mg mL−1, LOD (EPA-Me) = 0.0004 mg mL−1, LOQ (EPA-Me) = 0.0013 mg mL−1, LOD (DHA-Me) = 0.0004 mg mL−1, LOQ (DHA-Me) = 0.0013 mg mL−1.
Quantitative studies of the performed reactions were done by multiple points calibration curves (six points) with commercially available standards. R 2 (ARA) = 0.999, (ARA-Me) = 0.999, (EPA-Me) = 0.999, (DHA-Me) = 0.999. Figures S4–S6 in Supplementary Information show the HPLC runs of PUFE-Me/mt-PUFA-Me isomers and the runs of the corresponding mt-PUFA after purification.
Photolysis experiments to prepare mt-PUFA-Me
The cis-trans isomerization of ARA-Me, EPA-Me, and DHA-Me (15 mM in i-PrOH) was performed by photolysis λ = 250–260 nm in the presence of β-mercaptoethanol (7.5 mM) in a micro photochemical reactor, as previously reported. The solutions were first degassed with N2 for 20 min and then irradiated for 5 min, to avoid the formation of di-trans and multi-trans isomers. After the elapsed time, the mixtures were evaporated under a vacuum.
Ag-TLC purification of mt-ARA-Me
The crude mixture of ARA-Me was purified via preparative TLC pretreated for 15 min with 5% AgNO3 /ACN solution and then dried at 120 °C for 1 h. TLC eluents: ARA-Me hexane/Et2O/AcOH 9:1:0.4, TLC was run two times. The desired spots were scratched off, Rf (mt-ARA-Me) = 0.375, the silica suspended in EtOH and filtered. Afterward, the solvent was evaporated under reduced pressure and the product was stirred vigorously in 5% ammonium hydroxide solution for 20 min. After the elapsed time, the mixture was extracted with hexane, the collected organic phases dried over Na2SO4, filtered, and evaporated under reduced pressure. The desired mt mixtures of ARA-Me were isolated (1.86 mg) with purity by GC >98.4%.
Semipreparative reverse phase HPLC for the purification of mt-EPA-Me and mt-DHA-Me
EPA-Me and mt-EPA-Me have RT in the HPLC method of 55.072 and 59.081 min, respectively. Thus, a 2 mg mL−1 solution of the crude mixture from the photolysis of EPA-Me was prepared and injected in sequence, collecting fractions from each injection between 57.5 and 62.5 min. MeOH was evaporated from the collected elutes and the aqueous residues were freeze-dried to isolate the pure mixture of mt isomers. 1.6 mg of mt-EPA-Me, purity by GC >99% (Figure S2 in Supplementary Information). DHA-Me and mt-DHA-Me have RT in the HPLC method of 76.844 and 80.103/83.557 min, respectively. Thus, a 1 mg mL−1 solution of the crude mixture from the photolysis of DHA was prepared and injected in sequence (10 µL injection), collecting fractions from each injection between 79.00 and 85.00 min. MeOH was evaporated from the collected elutes and the aqueous residues were freeze-dried to isolate the pure mixture of mt isomers; 1.6 mg of mt-DHA-Me, purity by GC >98.4% (Figure S3 in Supplementary Information).
Hydrolysis protocols for the synthesis of mt-PUFA
Alkaline: One milligram of ARA-Me was dissolved in 0.5 mL 1 M NaOH and 0.5 mL ACN and vortexed vigorously for 1 min. The reaction mixture was vortexed at room temperature for 16 h. After the elapsed time, the reaction mixture was acidified with 5 M HCl and extracted with Et2O, the organic phases dried over Na2SO4, filtered and the solvent evaporated under vacuum. Injected in HPLC after dissolving in MeOH.
Acidic: One milligram of ARA-Me was dissolved in 0.73 mL of ACN and 0.28 mL of 5 M HCl in water. The mixture was vortexed vigorously for 1 min. The reaction mixture was kept at 100 °C for 1 h. After the elapsed time, 2 mL of 1 M NaOH were added to the sample, 1 mL of acetone was added and the mixture was injected in HPLC-MS after dissolving in MeOH. Figures S4–S6 in Supplementary Information show the purity of the mt-PUFA.
Supplemental Material
sj-docx-1-chl-10.1177_17475198221090908 – Supplemental material for A convenient route to mono-trans polyunsaturated free fatty acids
Supplemental material, sj-docx-1-chl-10.1177_17475198221090908 for A convenient route to mono-trans polyunsaturated free fatty acids by Fabrizio Vetica, Anna Sansone, Carla Ferreri and Chryssostomos Chatgilialoglu in Journal of Chemical Research
Footnotes
Acknowledgements
The Fondazione Di Bella is gratefully acknowledged for the grant given to F.V. We are grateful to Prof. Atsushi Matsuzawa and Dr Yusuke Hirata of Tohoku University for helpful discussion on the importance of mt-PUFA.
Authors’ note
Dedicated to Prof. Alwyn Davies, a distinguished member of the free radical chemistry community, on the occasion of his 95th birthday.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.