Convergent routes to various six-membered carbocyclic architectures exploiting the unique radical chemistry of xanthates are described in this brief review. Three approaches are discussed. The first is the modification of existing cyclohexane building blocks, namely, cyclohexanones, cyclohexenones and cyclohexenes. The second deals with the construction of six-membered carbocycles by associating the chemistry of xanthates with classical ionic reactions, especially the Robinson annulation, the Michael addition and the Horner–Wadsworth–Emmons condensation. Finally, the third route is the formation of six-membered rings by direct six-exo and, but more rarely, six-endo cyclisation modes. Many of the complex structures presented herein would be tedious to obtain by more traditional methods.
The cyclohexane ring is perhaps the most common structural motif in natural products. This observation justifies the importance of such textbook reactions as the Robinson annulation,1 the Diels–Alder reaction,2 and, more recently, the ring-closing metathesis (RCM).3 The last allows the construction of rings of almost any size. The modification of existing cyclohexanes – for example, by alkylation or aldol reaction of cyclohexanones,4 or by organocuprate addition to cyclohexenones5 – is also a popular strategy that is complemented by the partial or complete reduction of aromatic derivatives, especially through the powerful Birch and related dissolving metal reductions.6–8 Radical cyclisations have not found widespread application for the synthesis of six-membered carbocycles, in contrast to cyclopentanes which are readily accessible by fast five-exo ring closures.9,10 Six-exo and six-endo cyclisations allowing the direct formation of cyclohexanes are relatively slow transformations (see Scheme 19 below) that cannot easily compete with other pathways open to the precursor radical, for example, premature hydrogen abstraction commonly observed with organotin hydrides. We discovered some years ago that xanthates and related dithiocarbonyl derivatives (namely, dithiocarboxylates, trithiocarbonates and dithiocarbamates) allow the generation and capture of radicals by a unique mechanism that largely overcomes the problem of slow kinetics,11 a feature that can be exploited to build functional cyclohexanes by various ways summarised in the present review (for an account of the discovery of this process, see Zard12). This technology complements more classical routes and provides in many instances structures not easily accessible otherwise. However, before proceeding with a description of the synthetic approaches, it is necessary to discuss briefly the general features of the degenerative addition-transfer of xanthates.
As outlined in simplified form in Scheme 1, the radical chain reaction of xanthates 1 with alkenes 3, initiated, for example, by a peroxide, gives adducts 6.13–15 This process exhibits several features that make it unique. The active radicals – namely, R• and 4 – are reversibly stored in the form of mostly unreactive adducts 2 and 5. This increases significantly their effective lifetime, even in a concentrated medium, and, at the same time, lowers considerably their absolute concentration, thus limiting unwanted radical–radical interactions. Furthermore, the relative concentration of radicals R• and 4 is regulated through the fast equilibrium proceeding through intermediate 5 in a fashion that reflects their relative thermodynamic stabilities. To drive the process towards the desired product 6 and limit at the same time the formation of oligomers by further addition of radical 4 to alkene 3, this equilibrium must be biased so as to favour R• over radical 4. In other words, the partners must be chosen such that initial radical R• is more stable than adduct radical 4. This requirement neglects in a first approximation the influence of polar effects, which can be important and even decisive in some instances. In cases where the Y substituent on the alkene is electron donating, it is sometimes possible to oxidise adduct radical 4 into the corresponding cation 7 by electron transfer to the peroxide, and therefore move from a radical to an ionic manifold. There are many subtle features attached to this chemistry that cannot be discussed herein and the interested reader is directed to relevant literature for a more comprehensive discussion.16,17
Mechanism of addition of a xanthate to an alkene.
The ability to accomplish intermolecular additions of variously functionalised xanthates to even electronically unbiased alkenes, as well as the possibility of performing difficult (i.e. kinetically relatively slow) cyclisations or fragmentations opens up numerous synthetic possibilities not easily feasible by other methods. Several thousand additions have been realised using hundreds of different xanthates. The present review will describe the various ways this chemistry can be used to access six-membered carbocycles. These can be divided into three main strategies that are not necessarily independent of each other: (1) the modification of existing cyclohexanes, (2) the creation of six-membered ring by associating the xanthate addition with another more classical reaction and (3) the direct formation of the ring by radical cyclisation.
Modification of existing cyclohexanes
The modification of cyclohexanones and cyclohexenones is the most synthetically versatile. A few transformations involving xanthate 8 derived from cyclohexanone itself are displayed in Scheme 2. These reactions are mostly initiated with dilauroyl peroxide (or DLP; also known as lauroyl peroxide, Laurox® or Luperox LP®). Additions to allyl trimethylsilane and the acetal of acrolein lead to adducts 9 and 10, respectively.18,19 The xanthate group was reductively removed from the latter using tributylstannane to give simpler derivative 11. Other tin-free methods for reductively removing the xanthate group will be presented later. Notice, however, that this route to compound 11 (and indeed all the transformations in Scheme 2 and in several later schemes) corresponds to a formal alkylation of cyclohexanone enolate. In fact, radical additions of α-keto radicals generated via xanthate chemistry are complementary to enolate alkylations and in many ways even superior.20 Furthermore, regarding compounds 10 and 11, they are masked 1,5-ketoaldehydes which could be subjected to an internal aldol reaction1 or converted into pyridines by reaction with hydroxylamine or ammonium acetate under acidic conditions.21–24 For a review on the application of xanthates to the synthesis of pyridines, see Zard.25
Modification of cyclohexanone.
Addition to N-allylaniline 12 furnishes xanthate 13 which, upon further exposure to the peroxide, is converted into indoline 14.26 The peroxide is required in stoichiometric amounts to oxidise the cyclohexadienyl radical produced in the attack on the aromatic ring. This oxidation generates the corresponding cyclohexadienyl cation (see 7 in Scheme 1), which then aromatises by loss of a proton, as in the final steps of electrophilic aromatic substitutions, epitomised by the textbook Friedel–Crafts reaction. Hence, the need for stoichiometric amounts of peroxide. This sequence illustrates the use of the xanthate group to create two carbon-carbon bonds. In the present case, treatment of indoline 14 with cold sulfuric acid induces elimination of methanesulfinic acid resulting in aromatisation into indole 15. It is necessary to have electron-donating groups on the aromatic ring for this transformation to succeed (a methoxy group in this case).
Elimination of a methanesulfonyl radical has been exploited in an addition-fragmentation sequence on enesulfonamide 16 to produce ultimately pyrrole 17 in moderate yield (yields in parentheses correspond to yields based on recovered starting material throughout this paper).27 The pyrrole ring is formed by spontaneous closure of the intermediate imine onto the ketone of the cyclohexanone. A better approach to pyrroles is outlined in Scheme 3. It involves addition to vinyl esters such as vinyl acetate or vinyl pivalate (piv = pivalate) which, in the case of tetralone 18, produces adduct 19. This compound is a masked 1,4-ketoaldehyde and its condensation with 2-furfurylamine gives rise to pyrrole 20 by what may be viewed as a variation on the venerable Paal–Knorr reaction.28 Interestingly, the sulfur of the xanthate can be used to construct thiophene 21 by heating adduct 19 with potassium iodide and acetic acid in a microwave oven.29 These syntheses of pyrroles and thiophenes are quite general and modular: the ketone, vinyl ester and amine (or ammonia) in the case of pyrroles can all be modified.30
Synthesis of pyrroles, thiophenes and dienes.
The sulfur in the xanthate adducts can be exploited in yet another fashion. This is shown starting from addition product 22. Aminolysis of the xanthate liberates a thiol which spontaneously cyclises on the ketone to give an intermediate hemi-thioketal (not shown). Treatment with trifluoroacetic acid without purification results in dehydration to give the dihydrothiophene and oxidation with mCPBA finally affords sulfone 23. By heating with DBU in refluxing toluene, this Δ2-sulfolane isomerises reversibly to the Δ3-sulfolane isomer (not shown) which undergoes a cheletropic loss of sulfur dioxide to give diene 24.31 This unusual route to dienes is also fairly general. It was, for example, extended, with minor variations, to the synthesis of silyl-substituted dienes such as 25.32
The precursor to diene 25 is produced by the radical addition of xanthate 18 to vinyl trimethylsilane. These same adducts can be used in a completely different manner, as shown for adduct 26 derived from α-xanthyl-cyclohexanone 8 (Scheme 4).33 This compound can be used to access allenols by taking advantage of another transformation of the xanthate group, namely, its replacement with a dichlorovinyl motif by reaction with vinylsulfone 27.34 This furnishes allylic silane 28, where the ketone can be protected as ketal 29. This allows now application of the Corey–Fuchs reaction35 and capture of the intermediate lithium acetylide with cyclobutanone 30 to yield alkynol 31. Finally, treatment with fluoride delivers the expected allenol 32. This strategy furnishes allenes and allenols not readily available by more classical routes and illustrates another case where the xanthate group has allowed the formation of two carbon–carbon bonds.
Synthesis of propargyl alcohols and allenols.
Another manner for modifying cyclohexanones combines the chemistry of xanthates with that of sulfoxides (Scheme 5).36 Addition of tetralone xanthate 18 to vinyl ethylsulfide gives the corresponding adduct 33 in high yield. Thermolysis in diphenyl ether furnishes a mixture of unconjugated and conjugated enones 34 and 35, with the latter dominating, being the thermodynamically more stable isomer. The elimination of the xanthate is a retro-ene, Chugaev-like transformation (the actual Chugaev elimination takes place on the oxygen side of the xanthate).37 It requires much higher temperatures than typical Chugaev reactions but proceeds nevertheless in good yield in the present case as it is somewhat favoured by an anomeric effect resulting from the interaction of the lone pair of the sulfur atom in the ethylsulfide with the σ* antibonding orbital of the C–S bond of the xanthate. It is not necessary to separate these isomers. Oxidation with periodate furnishes the corresponding diastereomeric sulfoxides (not shown), all of which are converted into vinyl carbinol 36 by heating with triphenylphosphine in toluene. Under these conditions, the vinylic sulfoxide derived from sulfide 34 is in equilibrium with the allylic sulfoxide derived from sulfide 35, which is able to undergo the 2,3-sigmatropic rearrangement of the Mislow–Evans–Braverman reaction.
Synthesis of a vinyl carbinol.
While the α-position is the most common and easiest site for placing the xanthate group in cyclohexanones (and ketones in general), it is also possible to attach the xanthate on the carbonyl carbon itself, as pictured in Scheme 6. This is accomplished by first converting cyclohexanone into a geminal acetoxy chloride by reaction with acetyl chloride and anhydrous zinc chloride, and then substituting the chloride by the xanthate salt to give compound 37.38 This xanthate adds to alkenes in the usual way, as demonstrated by additions to allyl trimethylsilane and allylcyanide to afford adducts 38 and 39. Upon treatment with DBU, the latter undergoes two successive eliminations, first of the xanthate and then of the acetoxy group to provide cyanodiene 40 in good yield.
Additions of 1-acetoxy-1-xanthyl-cyclohexane.
This family of geminal acetoxy xanthates offers numerous possibilities for the creation of carbon–carbon bonds and clearly deserves to be explored further. Another equally interesting location for placing the xanthate is β to the carbonyl group.39 This installation can be achieved by Michael addition of xanthic acid to α,β unsaturated ketones, as shown for 3-methyl-cyclohexenone 41 in Scheme 7. The xanthic acid is generated in situ and captured as it is formed by treatment of the xanthate salt with acid. The temperature is kept low to avoid premature decomposition of the relatively unstable xanthic acid and the medium is kept acidic to prevent a retro-Michael fragmentation. Protection of the ketone as ketal 42 ensures no elimination of xanthic acid occurs during the radical addition. This is not always necessary, but β-xanthyl cyclohexanones are particularly prone to elimination by the retro-Michael process and masking of the ketone is a prudent move.
Radical addition at the β-position of cyclohexanones.
Compounds 43–46 are typical examples of radical additions (Scheme 7). In the case of the last derivative, the xanthate was exchanged for a chlorophenylthiyl group by reaction with the o-chlorophenyl disulfide.18 This reaction leading to 47 is not a chain process and requires stoichiometric amounts of peroxide. Oxidation to the sulfoxide, thermal elimination and deprotection with acid furnishes alkenylsilane 48. The regioselectivity of the sulfoxide elimination is directed by the silane.40 This is a little appreciated observation that has interesting theoretical and synthetic implications. For instance, treatment of compound 48 with NIS gives vinyl iodide 49 that smoothly participates in a Sonogashira coupling with phenylacetylene to yield enyne 50.
Modification of cyclohexenes is a further useful route to functional six-membered carbocycles. Direct addition to cyclohexenes is possible but can be complicated by variable and often low yields, regiochemical issues and inhibition through abstraction of allylic hydrogens. The modest yield of 52 from the addition of S-p-bromophenacyl xanthate 51 to cyclohexene itself is representative (Scheme 8).41 The case of norbornene and related derivatives is however special. The alkene is strained and therefore more reactive, and the hydrogens remaining in the allylic position have lost their allylic characteristics. They are not easily abstracted, thus eliminating a major source of trouble. Examples of additions to norbornene using an assortment of xanthates are displayed in Scheme 8. The first concerns the addition of xanthate 53 to give adduct 54 bearing a masked amine. Xanthate 53 is a nicely crystalline solid, inexpensive, and trivial to prepare and store.42 It is a very convenient reagent for preparing primary amines protected as phthalimides. Xanthates 55–60 were prepared in a similar manner allowing access to esters (55; in this case, initiation was accomplished by a redox process using an iridium complex catalyst),43 protected aminoacids (56),44 dichloroketone (57),45 protected α-ketoaldehyde (58),46 trifluoromethylketone (59),47 and a cyanohydrin benzoate (60).48 The last is a latent aldehyde. The diastereomeric ratio is variable and depends in part on steric factors. For instance, compound 59 containing a bulky O-neopentyl is formed as only the trans isomer. Another factor is the reversibility of the xanthate exchange (see Scheme 1), which tends to lead to the thermodynamic mixture. The reaction time can thus play a role in determining the observed diastereomeric ratio.
Functionalisation of cyclohexene and norbornene.
In contrast to intermolecular additions, cyclisations onto cyclohexenes are, not surprisingly, much easier to accomplish. Xanthates nevertheless allow cyclisations not readily performed by other methods because of the increased lifetime imparted to radicals generated through this chemistry. One case in point is the formation of γ-lactones from allylic alcohols by direct cyclisation. Such ring closures could not be accomplished starting from allylic bromoacetates using tributyltin hydride because of unfavourable rotamer distribution.49 Indeed, this difficulty forced the groups of Stork and Ueno to carry out the cyclisation on bromoacetals, which were then oxidised to the corresponding lactones.50–52 Using xanthates, cyclisations leading directly to lactones become trivial, as indicated by the two examples in Scheme 9(a). Thus, acetate and propionate derivatives 61 and 62 are readily converted into lactones 62 and 63 without need for high dilution.53 Furthermore, the xanthate groups in both products could be easily converted into the corresponding bromides by reaction with ethyl 2-bromoisobutyrate.
Functionalisation of cyclohexenols by intramolecular additions.
Stork and Nishiyama independently developed the use of silicon tethers to create carbon–carbon bonds in a stereocontrolled manner starting from cyclic allylic alcohols.54–57 The ability of xanthates to mediate additions onto unactivated alkenes allows a powerful expansion of this strategy through the use of vinylsilyl derivatives of allylic alcohols. Two series of such transformations are displayed in Scheme 9(b) and (c).58 Intermolecular radical addition to vinylsilyl-protected cyclohexenol 67 is followed by cyclisation to give bicyclic product 68, where three new chiral centres have been created with almost complete control of the (relative) stereochemistry. Reductive removal of the xanthate group using Barton’s hypophosphorus salt method59,60 furnishes the simpler derivative 69. In turn, the silicon group can be removed with fluoride to give cyclohexanol 70 or oxidised by the Tamao–Fleming protocol61,62 and the resulting diol protected to provide compound 71.
The addition of xanthate 53 to vinylsilyl-protected cyclohexanol 72 leads in a similar manner to the corresponding bicyclic product 73 (Scheme 9(c)).58 In this case, the xanthate was then used to introduce an allyl group by addition-elimination to allyl phenylsulfone to give compound 74.63 The roof-shape of the molecule ensures that allylation takes place from the least hindered exo-face. Thus, three new carbon–carbon bonds have been created, two of which in an intermolecular fashion.
The possibility of forming six-membered rings can also be exploited to modify cyclohexenes, as illustrated by the transformations in Scheme 10. The sequence in Scheme 10(a) is part of a formal synthesis of (±)-lepadin.64 Cyclisation of xanthate 75 gives rise to bicyclic ketone 76 which, after protection, affords ketal 77. The presence of an epimeric mixture is of no consequence, since both xanthates are precursors to the same radical, and vinylation with methyl styrylsulfone34 results in the formation of the desired exo isomer 78 as the major product. A similar cyclisation converts xanthate 79 into lactam 80, which could be easily processed into a berbane alkaloid (Scheme 10(b)).65 It is worth pointing out that both cyclisations in Scheme 10 proceed by an exclusive cis-mode to furnish the thermodynamically less stable, but perhaps synthetically more valuable ring junction.
Functionalisation of cyclohexenes using nitrogen substituents.
Cyclohexanes by a combination of radical addition and ionic cyclisation
Radical processes are generally tolerant of polar groups that often do not need prior protection. This property eliminates wasteful protection–deprotection steps and, importantly, allows the introduction of functional groups that can later be made to react together to create ring structures, and especially cyclohexanes. Xanthate chemistry is particularly apt at taking advantage of this feature by virtue of its ability to mediate intermolecular additions to electronically unbiased alkenes. One such strategy is illustrated in Scheme 11.66 The addition of α-xanthyl cyclobutanone 81 to alkene 82 leads to the expected adduct 83, where a ketone and a phosphonate have been placed in suitable relative positions to participate in a Horner–Wadsworth–Emmons condensation. Treatment with sodium hydride in dry THF at room temperature thus furnishes tricyclic derivative 84 without complications from the xanthate, which can be reductively removed or used to introduce other functional groups through ionic or radical processes. Cyclohexenes 85 and 86 were prepared in a similar manner.
Synthesis of cyclohexenes by the intramolecular Horner–Wadsworth–Emmons condensation.
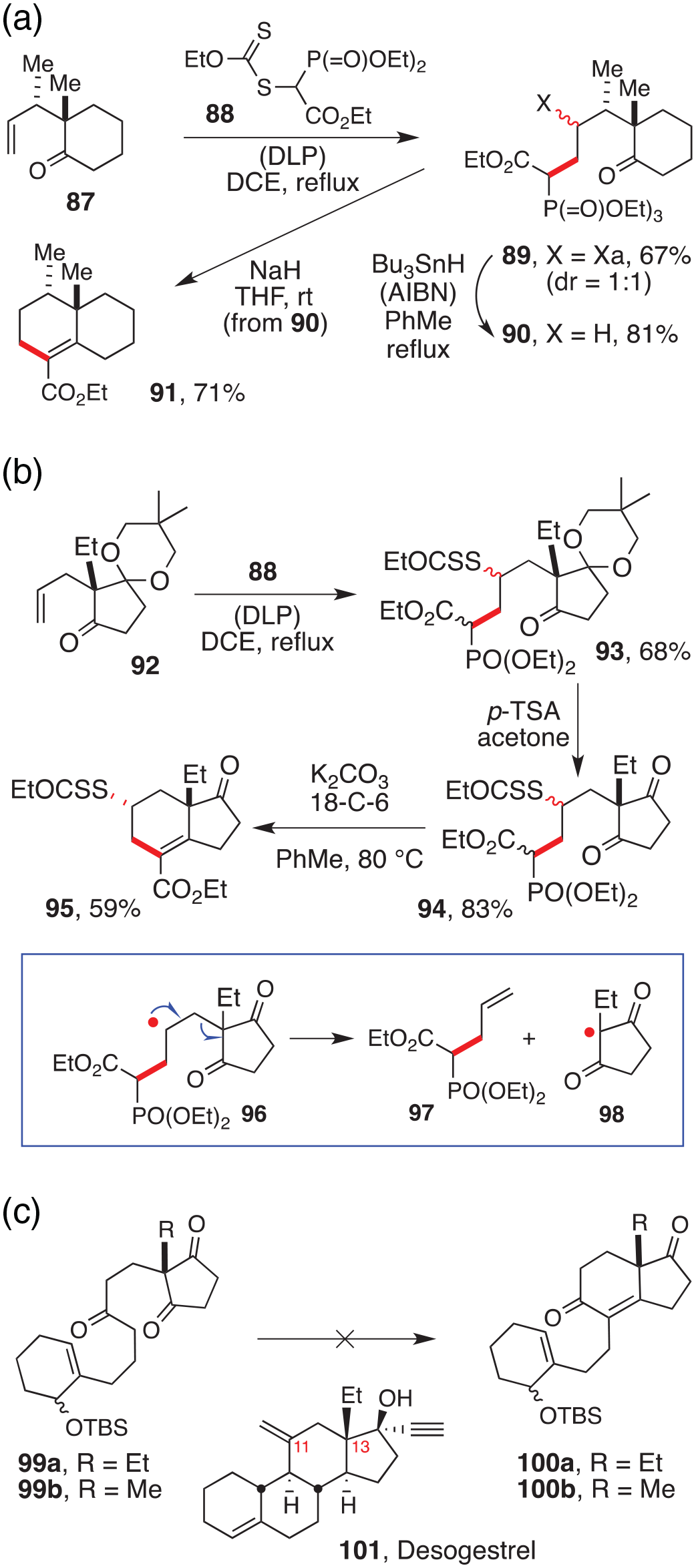
This route is obviously not limited to cyclobutanones and can be easily extended to other cyclic or open-chain ketones. Moreover, the positions of the ketone and the phosphonate can be switched, with the xanthate placed on the phosphonate partner and the alkene attached to the ketone. This alternative is highlighted by the examples in Scheme 12.67 Ketone 87 is readily obtained from 2-methylcyclohexanone and crotyl alcohol through the powerful Claisen [3,3]-sigmatropic rearrangement (Scheme 12(a)). Its reaction with xanthate 88 leads to adduct 89 where, again, the phosphonate and the ketone are suitably located to furnish a cyclohexene upon condensation. This was accomplished by first reducing off the xanthate with tri-n-butylstannane and exposing the product, 90, to the action of sodium hydride to give cyclohexene 91. In a similar manner, ketone 92 was converted into bicyclic cyclohexene 95 embodying the CD hydrindane portion of desogestrel 101, an extremely potent contraceptive (Scheme 12(b) and (c)).67 The radical addition first affords adduct 93, which is first deprotected before inducing the cyclisation with sodium hydride. The cyclisation of diketone 94 is interesting in that it takes place on the ketone that results in the xanthate group occupying the equatorial position (Scheme 12(b)). Thus, only one diastereoisomer is formed in the process.
Further syntheses of cyclohexenes by the intramolecular Horner–Wadsworth–Emmons condensation.
The need to use the monoprotected diketone is to avoid the unwanted fragmentation of intermediate radical 96 to give alkene 97 and radical 98, which was observed when starting with the unprotected diketone 94 (not shown). This fragmentation is favoured by the high stability of radical 98 that is delocalised over two ketone carbonyls. Mono-protection hence slows down considerably the rate of this unwanted fragmentation, since the resulting radical (i.e. monoketal of 98, not shown) is much less stable. The monoprotection offers another potential advantage, namely, the possibility of using a chiral diol to selectively protect one of the two enantiotopic ketones, and thus control ultimately the absolute configuration of the resulting bicyclic structure.68 It is further worth noting that, in their approach to desogestrel 101, Corey and Huang could not accomplish the Robinson annulation on compound 99a to access tetracycle 100a, despite extensive efforts, even though the same process in the methyl series 99b and 100b proceeded smoothly (Scheme 12(c)).69 The seemingly innocuous replacement of a methyl by an ethyl group completely shuts down the desired Robinson annulation in these compounds, in contrast to the intramolecular Horner–Wadsworth–Emmons condensation of the xanthate-derived precursor 94. Notice, furthermore, that the xanthate group in product 95 occupies the position corresponding to C-11 in desogestrel 101 (and steroids, in general) allowing the necessary subsequent modification of this key position. Carbon-11 is oxygenated in the clinically indispensable corticosteroids (cortisone, cortisol and their synthetic analogues), so that the strategy depicted in Scheme 12(b) could in principle be extended to this family of steroids.
In a third variation, the xanthate and the phosphonate are placed on either side of a ketone as in compound 102 (Scheme 13).70 This is a very interesting conjunctive reagent, since the xanthate can be used to accomplish intermolecular additions to alkenes and the α-keto-phosphonate used to perform Horner–Wadsworth–Emmons condensation on internal or external ketones or aldehydes. An illustration is provided by its addition to 2-allylcyclohexanone to give the corresponding adduct 103, from which the xanthate is removed with tri-n-butylstannane. Treatment of the resulting product 104 with base did not induce the expected condensation to give octenone 105, but, instead, triggered an internal aldol and afforded decalin 106. Without purification, this compound was reacted with benzaldehyde under mild basic conditions to give finally enone 107 in high overall yield. This substance is interesting because its dehydration would furnish a cross-conjugated dienone 108. Such dienones undergo interesting photochemical transformations and are substrates for the highly useful Nazarov cyclisation.71,72
Application of a radical-ionic conjunctive reagent.
Association with the Robinson annulation open several powerful routes to cyclohexenones. The most direct starts with tertiary xanthates derived by Michael addition of xanthic acid to enones, discussed above in connection with Scheme 7. As outlined in Scheme 14(a), xanthates 111 and 112 are easily prepared by addition of xanthic acid to enones 109 and 110. Their addition to vinyl acetate provides adducts 113 and 114, respectively.39 The geminal acetoxy xanthate motif is in fact a masked aldehyde (see adduct 19 in Scheme 3), which can be liberated by heating with acid in refluxing wet THF. Under these conditions, the internal aldol and dehydration occur spontaneously to furnish cyclohexenones 115 and 116 in good yield. A similar sequence starting with enone 117 affords spirocyclohexenone 120 via intermediates 118 and 119 (Scheme 14(b)).
An alliance with the Robinson annulation.
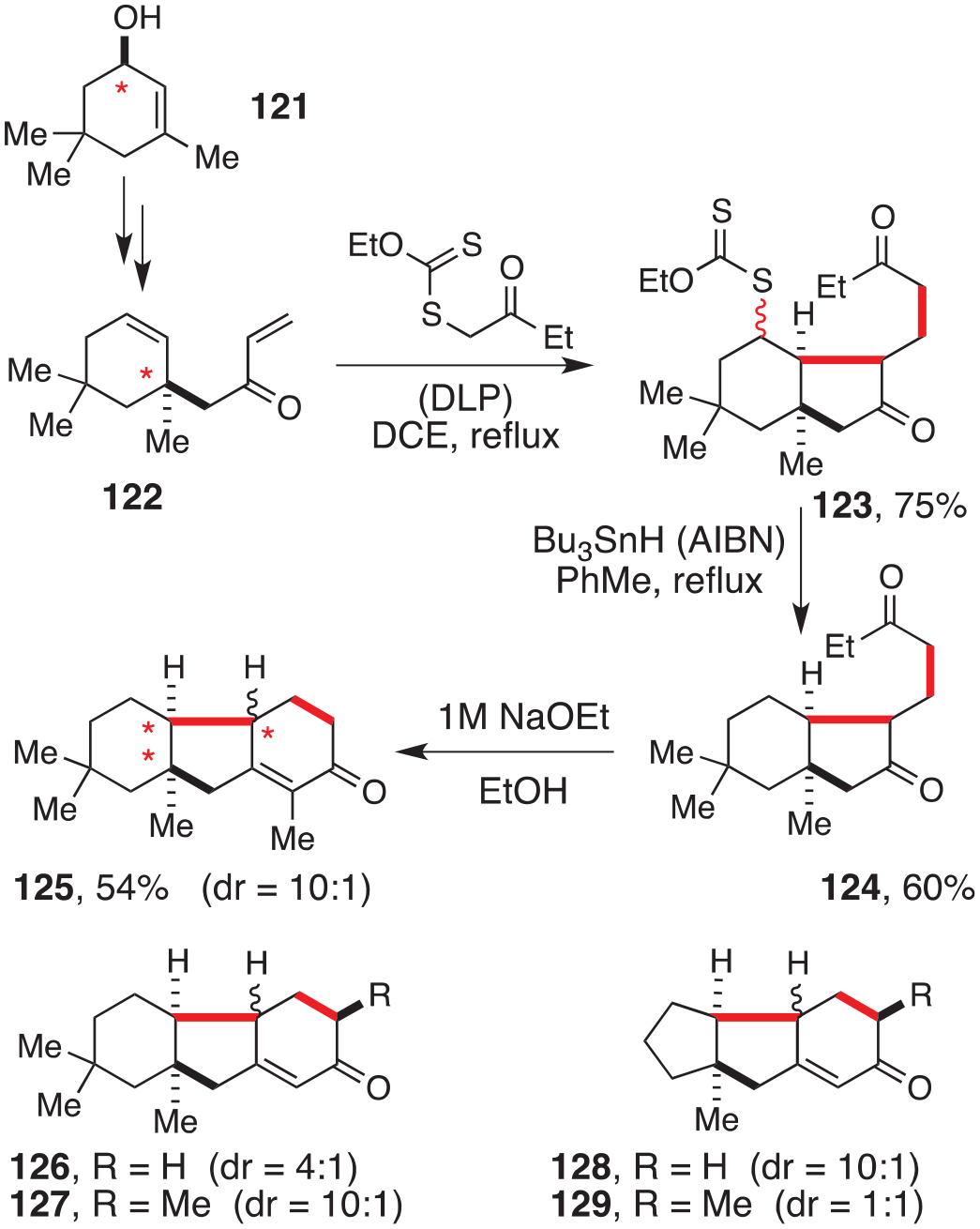
A more potent synthetic strategy arrays xanthate chemistry not only with the Robinson annulation but also with the Claisen rearrangement, as pictured in Scheme 15.73 The last reaction allows the conversion of isophorol 121 to enone 122, with complete suprafacial transfer of the stereochemical information of the alcohol in the former to the quaternary centre in the latter (starred carbons). The subsequent addition of the xanthate to the enone is followed by cyclisation to give bicylic intermediate 123, where two new C–C bonds have been created. Reductive removal of the xanthate group to simplify the structure and base-induced Robinson annulation produces tricyclic compound 125 with good control of the relative stereochemistry. Note that the chiral centre in the cyclohexanone subunit can be epimerised and corrected if needed through the extended enol or enolate. This expedient construction of polycyclic structures is modular and highly flexible, as indicated by the synthesis of compounds 126–129 by simply modifying the xanthate partner or the starting enone.
An expedient route to polycycles.
A less intuitive route to polycyclic architectures, also based on a combination of xanthate chemistry with the Claisen rearrangement and the Robinson annulation, proceeds through the generation and cyclisation of propargylic radicals (Scheme 16).74 Enyne 130 is prepared from 2-cyclopenten-1-ol using the suprafacial Claisen rearrangement as the key step. Addition of the acetonyl radical generated from xanthate 131 to the enyne side chain gives a propargylic radical that is captured internally by the cyclopentene to form allene 132. To simplify the structure, the xanthate group is again reductively removed. Solvolysis of the allenyl acetate motif in 133 in boiling aqueous acetic acid furnishes enone 134. Finally, DBU-induced Robinson annulation yields cyclohexanone 135.
Cyclisation of propargyl radicals.
Replacing the Robinson annulation with an internal Michael addition gives rise to a completely different skeleton, as shown in Scheme 17.74 Addition of malonyl xanthate 136 to the same enyne 130 proceeds in the expected manner to give allene 137. De-xanthylation to 138 and solvolysis of the allenyl acetate generates enone 139. Treatment with DBU now forms a cyclohexanone by conjugate addition of the malonyl anion to the enone and produces fused tricyclic derivative 140. Analogue 141 was prepared by an identical sequence. In both compounds 140 and 141, the configuration of the carbon bearing a methyl group next to the ketone can be modified via the enol or enolate, so that all the chiral centres can be controlled in principle. Propargyl radicals have very seldom been used in synthesis, yet they hold much promise since they allow a simple access to alkynes and allenes that cannot be readily obtained by more classical routes (note, for instance, that the allenes in Schemes 16 and 17 are tetrasubstitued).75,76
An alternative application of propargyl radicals.
In a project aimed at exemplifying a novel thermal transformation of allylic nitro compounds, xanthate chemistry was combined with an internal Henry reaction to construct a decalin structure (Scheme 18).19 Many years ago, it was found that ethylenediamine and a few of its congeners were effective catalysts for the Knoevenagel condensation of nitromethane with even hindered or unreactive ketones, such as 17-ketosteroids and tetralones.77–80 Thus, the ethylenediamine catalysed condensation of nitromethane with cyclohexanone 11, obtained using the xanthate addition as portrayed above in Scheme 2, furnishes nitroalkene 142 as mixture of two regioisomers. This is of no consequence, since the mildly basic conditions needed to mediate the Michael addition to methyl acrylate are sufficient to equilibrate the two isomers, resulting in the clean formation of compound 143. Deprotection gives nitroaldehyde 144 and sets the stage for the Henry addition, which is induced by DBU to furnish nitrodecalin 145 after acetylation of the alcohol. Heating this compound in refluxing o-dichlorobenzene in the presence of DABCO induces a [2,3]-sigmatropic rearrangement of the allylic nitro group and the resulting allylic nitrite (not shown) is hydrolysed in situ to provide allylic alcohol 146. The role of DABCO is to protect the allylic alcohol against dehydration under the harsh thermal conditions.
An association with the Henry reaction.
Cyclohexanes by direct radical cyclisation
Xanthates enable intermolecular radical additions, especially to electronically unbiased and unreactive alkenes, that cannot be accomplished by most other radical-based methods. This feature was exploited to bring together various functional groups that can then be made to react together to produce six-membered carbocycles, as underscored by the examples discussed in the foregoing section. The generation of cyclohexanes by direct ring closure is another route that is not readily implemented using traditional radical processes. The problem is the relatively slow rates of cyclisation as compared to other reaction modes open to the radical. This is in contrast to the easy synthesis of five-membered rings, where radical chemistry has had a dramatic impact.81–83 A glance at the prototypical rate constants displayed in Scheme 19 is sufficient to understand this difference.84 The five-exo closure of hexenyl radical 147 to cyclopentymethyl radical 148 is 50 times faster than the six-endo mode leading to cyclohexyl radical 149, even though the latter is thermodynamically more stable. The six-exo cyclisation of 150 into 151 is also sluggish and, moreover, in competition with the seven-endo closure to 152, which has a comparable rate, and the 1,5-hydrogen atom transfer (1,5-HAT) of the allylic hydrogen leading to radical 153.
Approximate rate constants for typical radical cyclisations.
The relative long lifetime of radicals generated through xanthate chemistry overcomes in many cases the problem of slow kinetics, and a proper choice of structures and substituents eliminates the six-exo/seven-endo competition as well as complications arising from 1,5-HAT. Thus, in xanthate 155, a substance efficiently obtained from enone 154, the seven-endo mode of closure is sterically blocked by the methyl groups, and the benzylic radical derived from the xanthate is too stabilised to abstract an allylic hydrogen (this is a nearly thermoneutral process).85 Treatment with peroxide indeed leads to the expected cyclohexanone 156 (Scheme 20). Bicyclic derivative 157 is another cyclohexanone obtained by a similar route. The cyclisation of xanthate 158 gives rise to cis-decalin 159 where an all-carbon quaternary centre has been created. In this case, 1,5-HAT is not possible and the seven-endo is mode would require going through a strained anti-Bredt type transition state.
Formation of cyclohexanones by direct cyclisation.
An alliance with the Diels–Alder reaction enables the synthesis of more complex structures, as outlined in Scheme 21.86 Ketone 160 is prepared by cycloaddition of methyl vinyl ketone with isoprene and may be obtained in enantiomerically pure form using chiral Lewis acid catalysts.68 Addition of ethoxyvinyllithium and bromination yields bromoketone 161, which is smoothly substituted with potassium O-ethyl xanthate. Exposure of the resulting xanthate 162 to initiation with DLP induces a clean cyclisation into cyclohexanone 163, isolated as a 1:1 mixture of epimers. This is of no consequence since both are progenitors of the same radical. Thus, reductive dexanthylation by the Barton method affords exclusively [3.3.1]bicyclononanone 164. Unlike the reversible transfer of the xanthate group, which tends to result in thermodynamic control, the transfer of the hydrogen from the hypophosphorus salt is irreversible and furnishes the kinetic product. In this case, it proceeds from the least hindered α-face to give the structure displayed.
Examples of an all-carbon temporary tether.
[3.3.1]Bicyclononanes are interesting scaffolds present in such natural products as garsubellin A and huperzine A.87,88 In the present case, treatment of the oxime derived from 164 with mesyl chloride and pyridine triggers an abnormal Beckmann rearrangement to give nitrile 165, where the acetyl side chain present in starting cyclohexene 160 has been regenerated. In this sequence, the acetyl group has thus served to direct the formation of two other chiral centres starred in blue. A more complex structure can be obtained using the xanthate in intermediate 163 to create another C–C bond by addition to acrolein diethyl acetal. Again, the addition takes place from the least hindered α-face to yield xanthate 166, which is easily reduced into compound 167. Abnormal Beckmann rearrangement of the corresponding oxime leads to nitrile 168 where, again, the acetyl group in starting cyclohexene 160 has enabled the control of two new chiral centres, one of which is quaternary. The special features of the xanthate addition-transfer have thus allowed the implementation of an all-carbon temporary tether strategy, proceeding by a six-exo closure instead of the easier five-exo cyclisation exploited in the classical Stork–Nishiyama and Stork–Ueno approaches. Compounds 169–171 are three other [3.3.1]bicyclononanes prepared in the same fashion but not subjected to the abnormal Beckmann rearrangement.
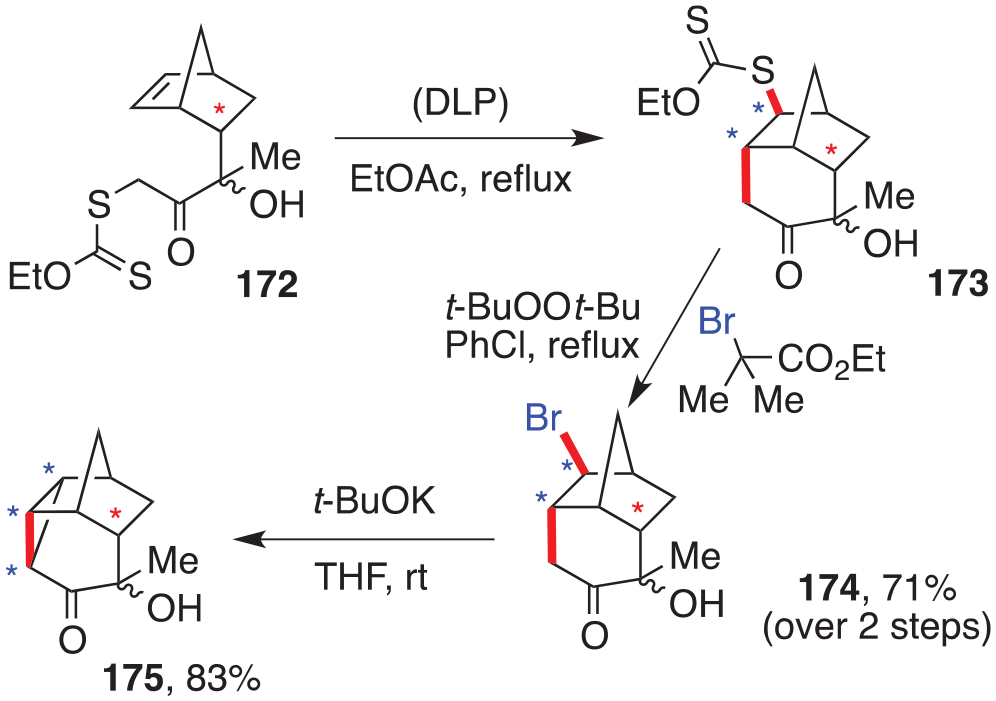
One further example illustrating this association of xanthate chemistry with the Diels–Alder cycloaddition is depicted in Scheme 22.86 It starts from xanthate 172, derived from the cycloaddition product of methyl vinyl ketone to cyclopentadiene. This compound cyclises smoothly into cyclohexanone 173 upon initiation with DLP, and the xanthate group is cleanly exchanged with a bromine to give 174. Finally, treatment with t-butoxide furnishes cyclopropyl ketone 175, where the red-starred chiral centre in the starting xanthate 172 allows control of the three blue-starred new chiral centres in the final product.
A cyclohexanone by direct cyclisation on a norbornene skeleton.
An alternative route to cyclohexanones exploiting the addition of ethoxyvinyllithium to enones is presented in Scheme 23.89 Thus, addition to cyclohexenone, bromination and substitution with xanthate provides compound 176. The radical derived from this xanthate does not readily cyclise, as this would lead to a strained cyclobutanone, but can be captured by an external alkene. In the case where this addition leads to a slightly stabilised radical, such as a tertiary or an oxygen stabilised radical, cyclisation onto the endocyclic alkene takes place concurrently to give directly decalinones 177, 178 and 179. When the adduct radical is secondary and not stabilised, as for addition to allyl trimethylsilane, the reaction stops at the normal addition product 180. Exposure of this substance to stoichiometric amounts of DLP in isopropanol induces both cyclisation and reductive removal of the xanthate group to give finally decalinone 181. The combination of stoichiometric peroxide in isopropanol is another convenient method for reducing xanthates.90 The hydrogen atom transfer from isopropanol is sufficiently slow to allow cyclisation to take place, even though the alcohol is used as the solvent. The transformations in Scheme 23 are the examples of a flexible, modular and versatile approach to highly decorated cis-decalinones, which are attractive building blocks for the synthesis of terpenes.
Formation of cyclohexanones by addition-cyclisation.
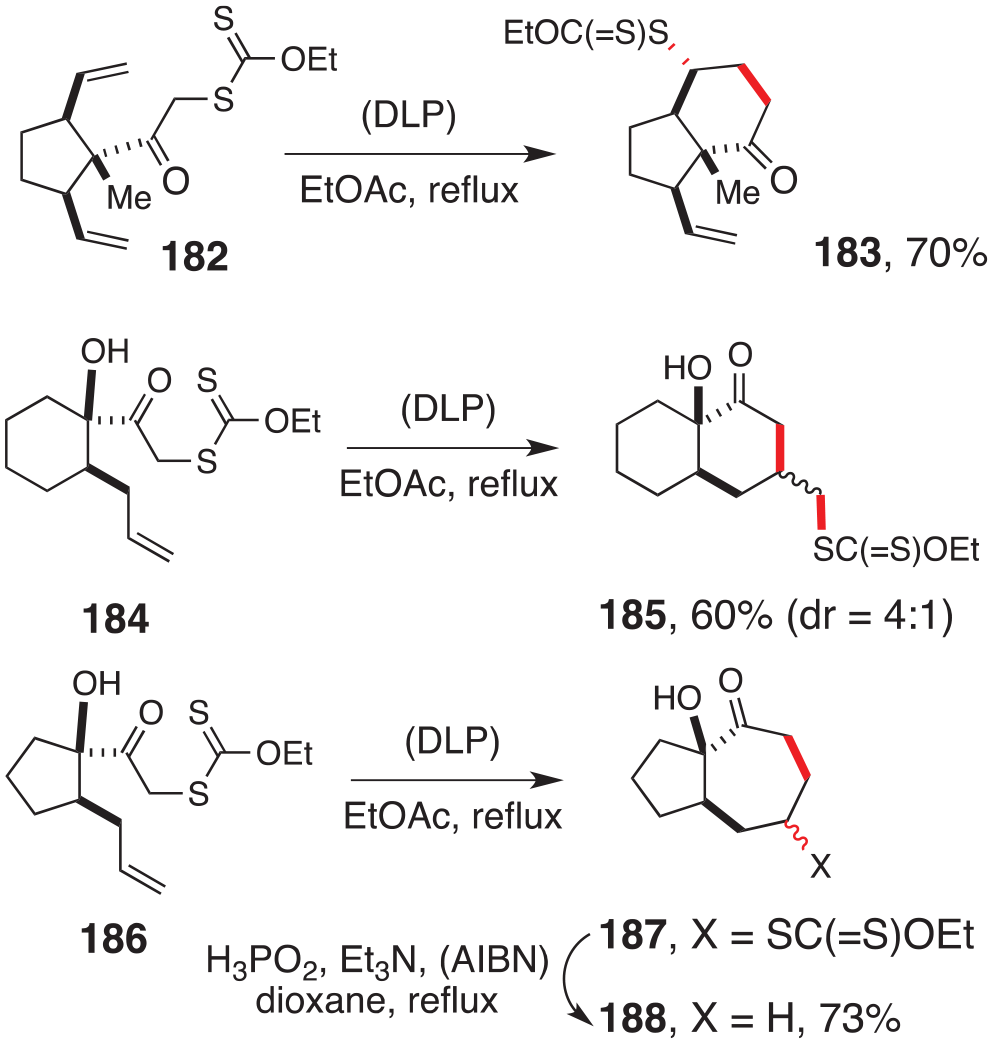
Accessing cyclohexanes by the six-endo ring closure mode is complicated by the strong and often overwhelming competition from the usually faster five-exo cyclisation. It is however possible to disfavour the latter cyclisation mode by introducing strain into the five-exo transition state, by sterically hindering the five-exo site of attack, or by introducing one or more sp2 carbons so that the larger bond angle favours the formation of the larger ring. These factors can, of course, be applied in combination, as illustrated by the examples in Schemes 24 and 25. The first transformation in Scheme 24, reported by Santelli and Ollivier,91 involves cyclisation of xanthate 182 to give trans-hydrindanone 183 by a six-endo ring closure. The alternative five-exo mode would have resulted in a significantly more strained trans-diquinane. The sp2 carbon of the ketone opens the bond angle (to 120°) and further favours the formation of the larger six-membered ring.
Further examples of cyclohexanones by direct by cyclisation.
A cyclohexanone from β-pinene.
These effects are also in play in the two other transformations in Scheme 4. In the first, there is no strain to impede the formation of trans-decalinone 185 from xanthate 184 by a six-exo cyclisation.89 This contrasts with the behaviour of xanthate 186, where the six-exo ring closure would have given rise to a somewhat strained trans-hydrindanone and the cyclisation proceeds preferentially by the seven-endo mode to give ketone 188, after reductive removal of the xanthate in the primary cyclisation product 187.
An interesting variation on this approach to cyclohexanones is displayed in Scheme 25 involving the reaction of di-xanthate 190 with β-pinene 189.92 The first addition-fragmentation produces xanthate 191, which then undergoes a six-endo cyclisation to give cis-decalinone 192. In this instance, this mode of cyclisation is favoured by the presence of the sp2 carbon of the ketone and, but perhaps to a lesser extent, the somewhat more congested trisubstituted extremity of the alkene which hinders the alternative five-exo closure.
Conclusion and further perspectives
The creation of C–C bonds is the backbone of organic synthesis. The degenerative addition-fragmentation of xanthates and related dithiocarbonyl derivatives is particularly powerful in this respect because, on one hand, it allows difficult (i.e. kinetically sluggish) intramolecular and, especially, intermolecular additions to activated and electronically unbiased alkenes and, on the other hand, it tolerates the presence of unprotected polar groups such as ketones, esters, carboxylic acids, alcohols, amides and phosphonates. This opens up numerous opportunities for the construction of a broad variety of molecular architectures through the alliance of xanthate chemistry with ionic, organometallic, pericyclic and photochemical reactions. One final example to further underscore the unique synthetic potential is pictured in Scheme 26. Addition of phenacyl xanthate 193 to butenyl acetate gives the corresponding adduct 194. Upon exposure to stoichiometric amounts of peroxide, this compound undergoes cyclisation onto the aromatic ring to produces tetralone 195. Replacement of the acetate with a tosylate and treatment with base affords tricyclic ketone 196. Finally, the Birch reduction and in situ methylation of the enolate furnishes dienone 197. A complex tricyclic structure has thus been expediently assembled by associating xanthate chemistry with the powerful alkylative Birch reduction. Even more complex structures should be accessible by starting with more substituted phenacyl xanthates and modifying the alkene partner and alkylating agent. Incidentally, the formation of tetralone 195, one of many examples,41 can be considered as an example of formation of a ‘cyclohexanone’ by an addition-cyclisation. In summary, while the present brief review has concentrated on only a narrow aspect of xanthate chemistry, being limited to six-membered carbocycles, it will hopefully provide an idea of the possibilities.
An alliance with the alkylative Birch reduction.
Footnotes
Acknowledgements
The author is greatly indebted to his co-workers, whose names appear in the references, for their skill, dedication and enthusiasm. The author is also most grateful to Dr Béatrice Sire, who, over a period of 25 years, contributed immensely to many of their projects and was instrumental to their success.
Author’s note
This paper is respectfully dedicated to Professor Alwyn Davies on the occasion of his 95th birthday.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The author also thank the following organisations and companies who have provided financial support over the years: Ecole Polytechnique, CNRS, DGA, MNRT, The China Research Council, Aventis (now Sanofi), Laboratoires Servier and Syngenta.
ORCID iD
Samir Z Zard
References
1.
JungME. Tetrahedron1976; 32: 3.
2.
NicolaouKCSnyderSAMontagnonT, et al. Angew Chem Int Ed2002; 41: 1668.