
Abstract
Lactol carbocyclisations provide a succinct method of constructing the oxabicyclo[3.2.1]octane scaffold, a motif present in various natural products of medicinal interest. Lactols containing an unsaturated ketone or ester were prepared by olefin cross-metathesis; an electrophilic alkene derived from methyl vinyl ketone underwent concomitant terminal α-methylenation and oxa-Michael addition to give a bridged lactol which then underwent oxygen-to-carbon transposition in the presence of titanium (IV) chloride giving the desired unsaturated carbocyclic seven-membered bridged ether via a novel dehydrative cascade considered to involve titanium enolates.
Keywords
A stepwise enone-lactol carbocyclisation is described. Concomitant α-methylenation and intramolecular oxa-Michael addition afford a bridged acetal which in the presence of TiCl4 undergoes oxygen-to-carbon transposition giving an unsaturated carbocyclic bridged ether.

Introduction
Bridged ethers are found in numerous natural products,1–7 including the potent anti-cancer agent englerin A (Scheme 1).2,5,6 Recently, oxabicyclo natural products containing the benzo-8-oxabicyclo[3.2.1]octane core (Figure 1) have been identified as Gram-positive antibiotics, anti-parasitic agents and anti-cancer agents,1,2 adding further incentive to access such bridged systems. Accordingly, general methods of constructing polysubstituted O-bridged systems with control of stereochemistry are valuable in the synthesis of natural products and new scaffolds for medicinal chemistry. To this end, we identified lactol carbocyclisations as an effective method of constructing bridged ethers, 8 including the trans-addition across an alkene terminus to give a bridged ether (Scheme 1, eq. i). When the π-nucleophile is an aromatic ring, these lactol carbocyclisations have also proved efficient,1,3,8–13 creating a benzenoid ring fused across the 2,3-positions of the bridged ether positions (numbering as in Scheme 1). Such methodology has become a cornerstone in the synthesis of various bridged-ether natural products.1,3,9–13
Representative oxabicyclo[3.2.1]octane natural products, a lactol carbocyclisation route to such bridged ethers, and a proposed enone–lactol–Nazarov cyclisation cascade.

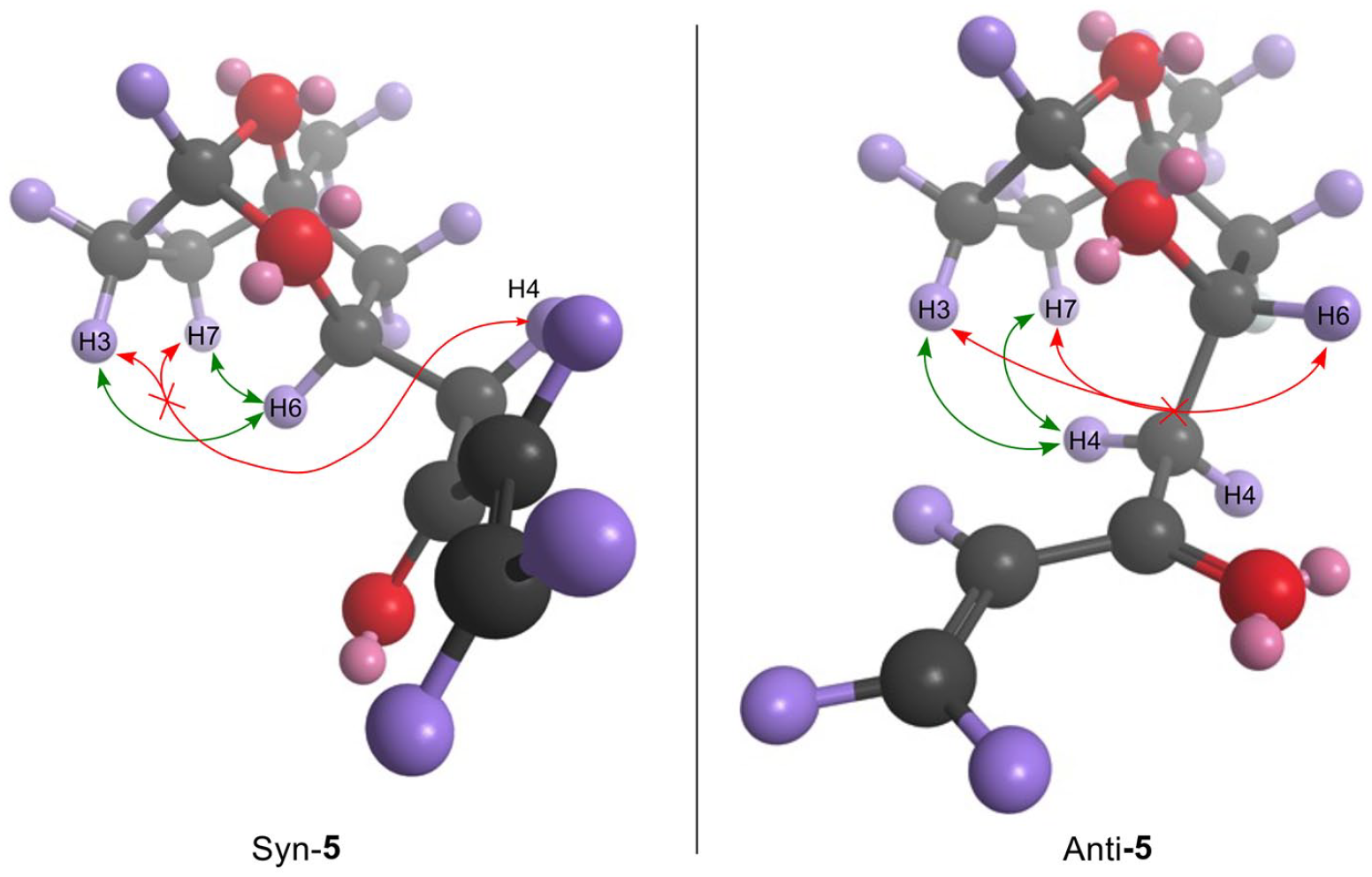
Comparision of syn-and anti-5 expected NOESY cross peaks.
Although aromatic termini serve effectively as a trap for the lactol-derived oxocarbenium ion, the scope of such lactol cyclisations (Scheme 1, eq. i) would be greatly increased if an alkene π-nucleophile could act as a charge relay, delivering carbocyclisation cascades. For example, the bridged perhydroazulene scaffold, present in englerin A and related natural products of therapeutic significance, might be constructed via a sequential lactol–Nazarov cyclisation (Scheme 1, eq ii). This raised the question of whether a direct lactol–enone cyclisation was feasible where R2 in lactol
Results and discussion
Addition of allylzinc bromide to methyl levulinate afforded the known lactol

Synthesis of enone lactol
Attention was then turned to the synthesis of dienone termini (Scheme 1, eq. ii) since those might first undergo Nazarov cyclisation
22
followed by lactol carbocyclisation induced by an enol intermediate (i.e. the reverse order of that in eq. ii). Accordingly, Mannich α′-methylenation studies were undertaken in the presence of diisopropylammonium trifluoroacetate, using Connell’s procedure.
23
α-Methylenation of
It was noted that the removal of alkene unsaturation through the formation of acetal
The constitution of enone acetal
In the same fashion, confirmation of the constitution of the dienone acetal
Confirmation of the constitution of dienone ether
The present observations can be accounted for by the mechanistic pathways outlined in Schemes 3 and 4. The combined yield of isolated products

Proposed mechanism for the formation of bridged enone acetals
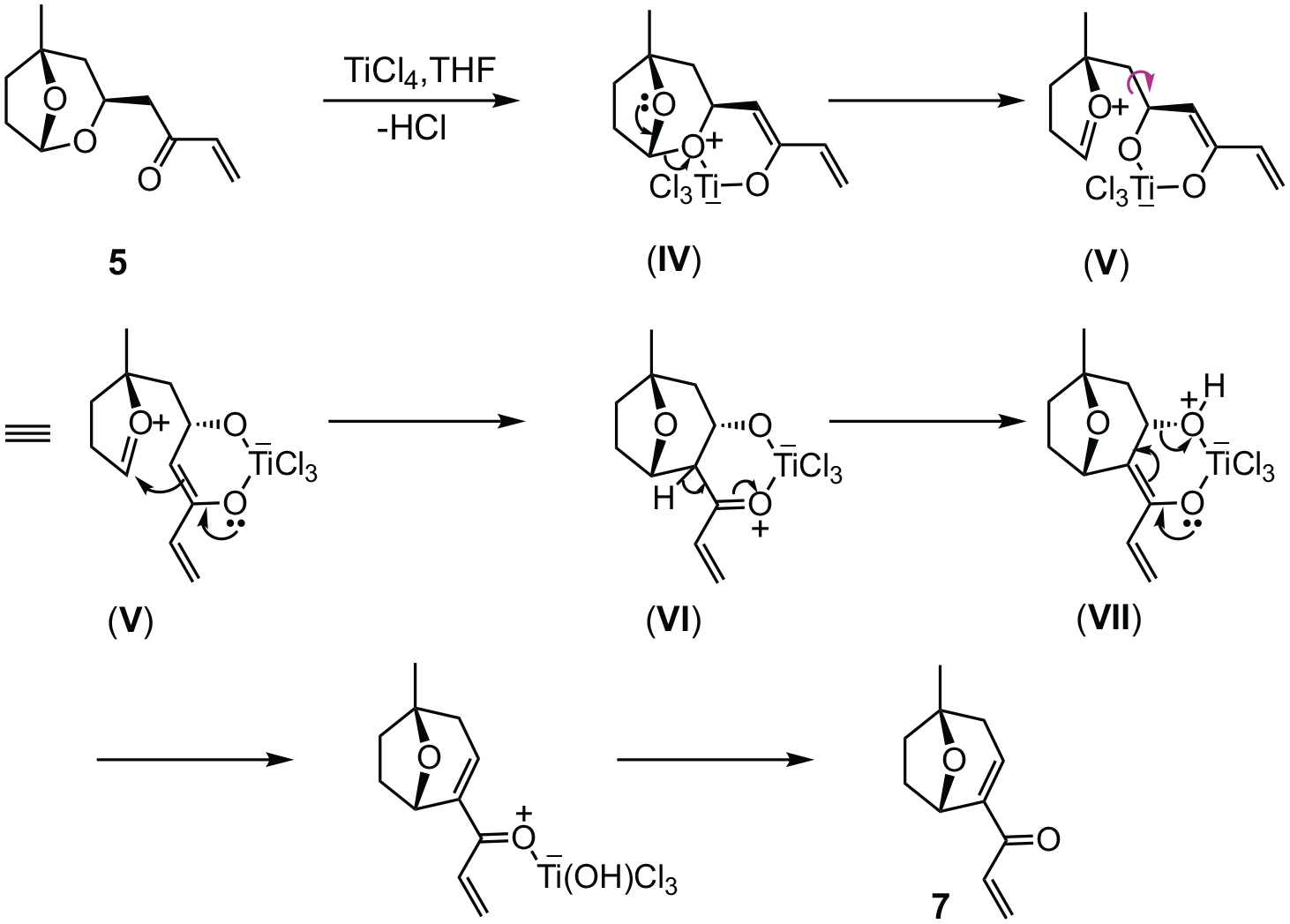
Proposed mechanism for the transposition of acetal
Chelated titanium enolates such as (
Key features identified in this study can be linked with inferred and known lactol formations to postulate a set of cascade processes from a totally acylic precursor (Scheme 5). A previous lactol carbocyclisation could only be readily accounted for by postulating a linear γ-hydroxycarbonyl compound as a key intermediate in the cascade rearrangement of certain 3,4-epoxy alcohols. For example, a bridged ether possessing a 6,7-fused cyclopentane ring (numbering as in Scheme 1) was formed in greater than 80% yield from a putative γ-hydroxyaldehyde.
8
Those observations and the present work confirm that conditions for lactol carbocyclisations are generally compatible with the conversion of a linear γ-hydroxycarbonyl compound such as
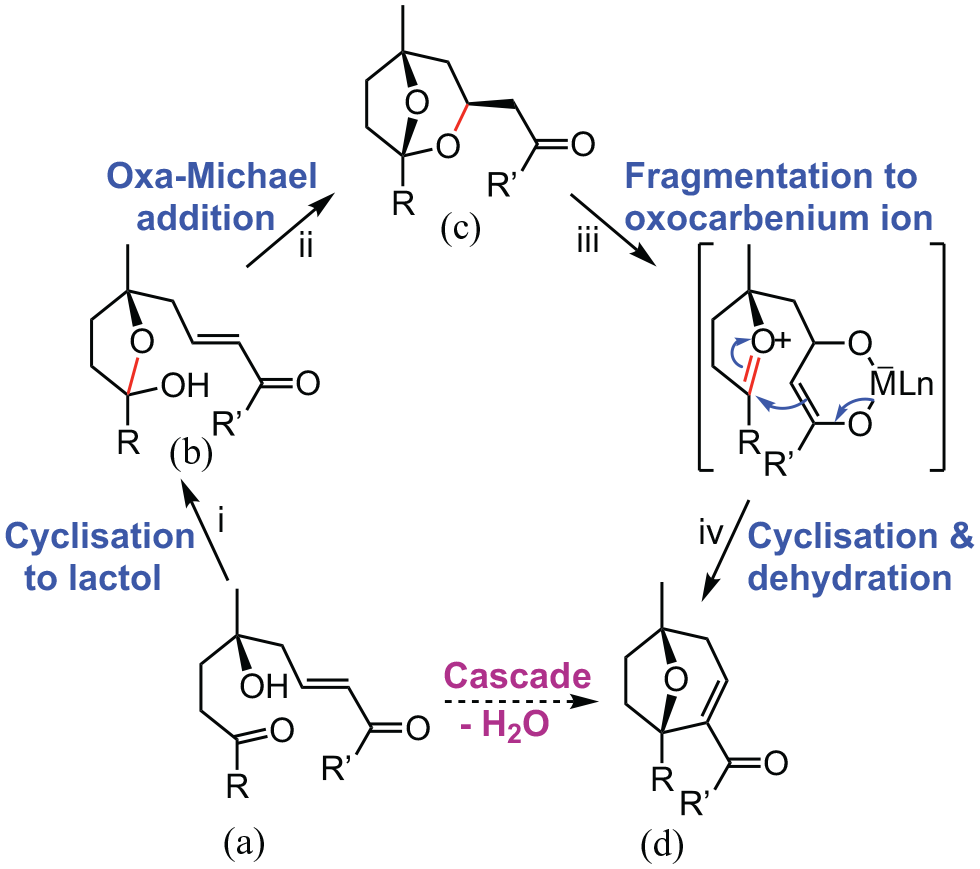
Assembly of identified steps into a proposed cascade from acylic reactants.
Conclusion
Hitherto, lactol carbocyclisations have been confined to the use of relatively electron-rich alkenes such as π-nucleophiles. This study has achieved a formal process corresponding to lactol carbocyclisation using an electrophilic alkene derived from methyl vinyl ketone. Under the conditions of terminal α-methylenation of methyl ketone, oxa-Michael addition of lactol occurred, affording a bridged lactol which then underwent oxygen-to-carbon transposition to give the desired carbocyclic bridged ether via a cascade inferred to involve titanium enolate species. In contrast to the powerful Ferrier type II27–29 and Petasis–Ferrier reactions,17,30–33 which involve trapping of oxocarbenium ions by enol ethers, both generated from atoms within the ring that is cleaved, the present oxygen-to-carbon transposition involves remote but concomitant generation of the oxocarbenium ion and its trapping by a presumed metal enolate derived from a ketone moiety external to the ring cleaved, a process which has not been previously reported, to the best of our knowledge. In addition to these novel transformations, and taken together with previous lactol-forming reactions, the results suggest a route from acyclic hydroxy ketones to the bridged ethers might be achieved without the isolation of acetal intermediates. This methodology enhances access to the 8-oxabicyclo[3.2.1]octane scaffold, one found in a range of natural products possessing medicinal properties.
Experimental section
General
Starting materials and reagents were purchased from commercial sources and used without further purification. Diisopropylammonium trifluoroacetate was prepared as previously decribed. 23 Anhydrous THF was purchased from Acros Organics. Anhydrous dichloromethane was produced by distillation over CaH2, stored over 4 Å molecular sieves and used within 1 week of distillation. Molecular sieves were washed with dichloromethane before use and dried at 180 °C for at least 48 h. All reactions were performed under an argon atmosphere and using glassware pre-dried in an oven (150 °C) and then cooled to room temperature under a nitrogen atmosphere. Flasks for reactions performed under an inert atmosphere were fitted with Suba·Seal rubber septa, sealed with Parafilm, and purged with nitrogen. Flash column chromatography was performed using Merck 0.040–0.063 mm, 230–400 mesh silica gel. Evaporation refers to the removal of solvent under reduced pressure. Thin-layer chromatography (TLC) was performed on Merck 0.2 mm aluminium-backed silica gel 60 F254 plates and visualised under UV light (254 nm) either by staining with alkaline potassium permanganate with subsequent heating, or by exposure to an acidic (1% v/v conc. H2SO4) ethanolic solution of vanillin (6% wt/vol). Alternatively, the TLC plate was developed in an iodine chamber. Infrared (IR) spectra were recorded on a Perkin−Elmer Spectrum One FTIR spectrometer; absorptions are quoted in wavenumbers. 1H and 13C NMR spectra were recorded on either a Bruker Avance III 400 MHz spectrometer or a Bruker Avance Neo 700 MHz spectrometer and calibrated using residual undeuterated solvent as an internal reference; chemical shifts are in parts per million (δ) and coupling constants (J) are given in Hertz (Hz). The following abbreviations were used in signal assignments: singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), and multiplet (m). Diastereoisomeric ratios were calculated from 1H NMR peak areas. High-resolution mass spectra (HRMS) were recorded using either an Agilent ESI TOF (time of flight) mass spectrometer at 3500 V emitter voltage or using a Waters LCT Premier QTOF spectrometer connected to a Waters sample manager 2777C at University College London.
Preparation and use of activated zinc
A heterogeneous solution of zinc (1.60 g, 24.4 mmol) in aqueous hydrochloric acid (5%, 20 mL) was stirred for 10 min or until effervescence had subsided. The resulting zinc slurry was then filtered, washed with water (2 × 5 mL), ethanol (2 × 5 mL) and then with diethyl ether (2 × 5 mL). In experiments involving the preparation of allylzinc reagents, the activated zinc was either used directly or oven-dried (180 °C) for at least 24 h. After allowing the washed zinc to cool, it was added to the stirred reaction mixture, and where necessary, the reaction was initiated by the addition of a single crystal of iodine, resulting in a significant exotherm which subsided after 5–10 min.
5-Allyl-5-methyldihydrofuran-2(3H)-one (2 )
According to a literature procedure,
20
modified by the addition of HMPA, a round-bottom flask was charged with a stirred suspension of oven-dried (180 °C) zinc powder (<150 μm, 99.995%) (5.71 g, 87.3 mmol) in THF (90 mL) under a nitrogen atmosphere. To the suspension was added methyl levulinate (4.32 mL, 34.9 mmol), followed by HMPA (0.61 mL, 3.5 mmol) and allyl bromide (7.53 mL, 87.1 mmol). The resulting dark grey suspension was heated to 45 °C. After 8 h, the reaction was complete, as indicated by TLC analysis. The dark grey suspension was evaporated and the residue was quenched with saturated aqueous ammonium chloride (100 mL). The aqueous layer was extracted with diethyl ether (3 × 100 mL) and the combined organic layers were washed with hydrochloric acid (1 M, 75 mL) and brine (100 mL), and then dried (MgSO4) and evaporated. The residue was purified by chromatography on silica gel using hexane: ethyl acetate (90:10 then 85:15, Rf = 0.17) to give
5-Allyl-5-methyltetrahydrofuran-2-ol (3 )
A solution of
Methyl (E)-4-(5-hydroxy-2-methyltetrahydrofuran-2-yl)but-2-enoate
To a stirring solution of
(E)-5-(5-Hydroxy-2-methyltetrahydrofuran-2-yl)pent-3-en-2-one (4 )
To a stirring solution of
One-pot α-methylenation-oxa-Michael cyclisation of 4 to give 1-methyl-3-(buta-1-en-3-one)-4,8-dioxabicyclo[3.2.1]octane (5 ) and 1-methyl-3-(2-(penta-1,4-dien-3-one))-4,8-dioxabicyclo[3.2.1]octane (6 )
A mixture of
For
For
Fragmentative carbocyclisation of 5 to give 1-methyl-4-(prop-3-en-1-one)-8-oxabicyclo[3.2.1]oct-3-ene (7 )
A stirring solution of
Supplemental Material
sj-pdf-1-chl-10.1177_17475198221079498 – Supplemental material for A stepwise lactol carbocyclisation to bridged ethers via a keto–acetal cascade
Supplemental material, sj-pdf-1-chl-10.1177_17475198221079498 for A stepwise lactol carbocyclisation to bridged ethers via a keto–acetal cascade by Sean McCarthy and Charles M Marson in Journal of Chemical Research
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.