Experimental details
General directions: Starting materials were supplied from commercial sources and used without further purification. All commercially available solvents were used as supplied unless otherwise stated. All ‘dry’ solvents were dried and distilled by standard procedures or were processed through a Grubbs type still. Oxygen-free nitrogen was obtained from BOCgases and was used without further drying. Infrared spectroscopy was performed on a Fourier-transform infrared(FTIR) spectrometer. Routine electrospray mass spectra and high-resolution mass spectra (HRMS) were run on electrospray ionisation (ESI). Chiral HPLC was performed on chiralpakIB, IC or AS-H columns as stated. The nuclear magnetic resonance (NMR) spectra were recorded at 25 °C on 300 or 400MHz spectrometers, as indicated. All peak assignments are confirmed by 2D experiments (
1
H-
1
HgCOSY, 1H-
13
CHSQC). TLC (thin layer chromatography) was performed on 60F254 aluminium-backed plates with realisation by ultraviolet (UV) irradiation and/or chemical staining. Flash column chromatography was performed with silica, particle size 0.040–0.063 mm.
Δ
1
-Piperideine 8:
68
Piperidine 6 (5.17 g, 60.72 mmol) was added to a solution of N-chlorosuccinimide (15.00 g, 112.33 mmol) in diethyl ether (400 mL). The solution was stirred at rt for 1h. After filtration and rinsing of the precipitate with diethyl ether (40 mL), the diethyl ether solution was washed with water (2 × 400 mL) and brine (200 mL) and then dried with MgSO4. The N-chloropiperidine solution was then filtered and concentrated under reduced pressure to roughly 80mL volume. The resultant solution was added dropwise to a solution of KOH (4.00 g, 71.20 mmol) in absolute ethanol (64 mL). During this process, the temperature was maintained between 5 °C and 10 °C. After addition, the mixture was stirred at room temperature for 24h and filtered. The precipitate was rinsed with absolute ethanol (60 mL), and the combined solution was concentrated to about 25mL. Diethyl ether (200 mL) and water (20 mL) were added and the mixture was extracted. The phases were separated and the aqueous phase was then further extracted with diethyl ether (2 × 100 mL). The combined organic phases were washed with brine and dried with anhydrous MgSO4, filtered, and concentrated in vacuo to afford 8 (2.5 g, 60%) as yellowish oil, which gradually solidified. This material was subsequently used without further purification and can be stored indefinitely at low temperature.
(−)-(2S)-(2-Oxopropyl) piperidine-1-carbamic acid benzyl ester (Cbz-pelletierine) 7:
68
Δ
1
-Piperideine 8 (Typical experiments used the solid 1 | -piperideine trimeric form of 8. However, the initially isolated oil form of 8 can also be used with similar results)67,82 (670 mg, 8.06 mmol, 1 eq.), acetone (27 mL, 367.59 mmol, 46 eq.), DMSO (27 mL), water (3.4 mL), and l-proline (185 mg, 1.61 mmol, 0.2 eq.) were mixed and stirred for 1 h at room temperature. An aqueous saturated solution of sodium bicarbonate (50 mL) was added to the mixture, which was extracted with CH2Cl2 (2 × 50 mL). The organic layer was washed with brine (50 mL), filtered and dried over anhydrous MgSO4. After filtration, the solvent was removed under reduced pressure, and the crude pelletierine was used immediately without purification [HRMS (ES) calcd for [(C8H15NO + H)]+ 142.1232, found 142.1232 (0.1 ppm)]. The residue obtained above was diluted in CH2Cl2 (25 mL) and a 0.4 M solution of aqueous sodium carbonate (18 mL, 7.20 mmol, 0.9 eq.) was added. The mixture was cooled to 0 °C and benzyl chloroformate (0.9 mL, 6.30 mmol, 0.8 eq.) was added dropwise. The ice bath was removed and the solution was stirred at rt overnight. The solution was diluted with CH2Cl2 (25 mL) and extracted. The resultant organic layer was washed with water (50 mL), and then with brine (50 mL), dried over anhydrous MgSO4, filtered and concentrated in vacuo. This gave the crude product which was purified by flash column chromatography (CH2Cl2/EtOAc; 6:1) to afford the title compound 7 (1.25 g, 72 % yield over two steps) as light-yellow oil. Rf = 0.55 (EtOAc/CH2Cl2; 1:6); [α]D
20
= –10.5 (c = 1.00, CHCl3) {lit.,
83
[α]D
28
= –10.0 (c = 0.50, CHCl3)}; HRMS (ES+) calcd. for [(C16H21NO3+Na)]+ 298.1491, found 298.1430 (+3.8 ppm); IR: 3032, 2938, 2862, 1688, 1497, 1353, 1258, 1165, 1049, 735, 697 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.42–7.32 (m, 5H), 5.11 (d, J = 12.5 Hz, 1H), 5.08 (d, J = 12.5 Hz, 1H), 4.84 (br. s, 1H), 4.07 (br. s, 1H), 2.87 (app. t, J = 12.5 Hz, 1H), 2.73–2.61 (m, 2H), 2.15 (s, 3H), 1.78–1.34 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 206.9 (CO), 155.3 (CO), 136.7 (C), 128.4 (CH), 127.9 (CH), 127.8 (CH), 67.1 (CH2), 47.5 (CH), 44.3 (CH2), 39.8 (CH2), 30.0 (CH3), 28.3 (CH2), 25.2 (CH2), 18.8 (CH2) ppm; HPLC analysis (Chiralpak IB), heptane/EtOH; 98:2 (1mL/min.): (−)-7 tr = 12.1 min., (+)-7 tr = 13.6; 90:10 e.r.
Quinazolinone synthesis: General Procedure: A mixture of the anthranilic acid (8.0 g, 0.06 mol) and formamide (10.8 g, 0.24 mol, 4 eq.) was stirred at 165 °C (oil bath temperature) for 4h. The mixture was cooled before water (15 mL) was added. At this stage copious solid material appeared. The mixture was then warmed to 60 °C and water (30 mL) was added. The mixture was stirred for another 30 min at 60 °C. The resulting precipitate was filtered and then recrystallised with ethanol to give the quinazolinone compounds 10a–10f.
6-Chloroquinazolin-4(3H)-one10a: 5-Chloroanthranilic acid 9a gave 10a as a white solid. Yield: 61%. M.p. 274 °C–275 °C (EtOH) {lit.,
77
259 °C–261 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 12.45 (br. s, 1H), 8.13 (s, 1H), 8.06 (d, J = 2.5 Hz, 1H), 7.85 (dd, J = 8.5, 2.5 Hz, 1H), 7.70 (d, J = 8.5 Hz, 1H).
6-Bromoquinazolin-4(3H)-one10b: 5-Bromoanthranilic acid 9b gave 10b as a white solid. Yield: 70%. M.p.267 °C–269 °C (EtOH) {lit.,
77
261 °C–267 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 8.17 (d, J = 2.5 Hz, 1H), 8.12 (s, 1H), 7.94 (dd, J = 8.5, 2.5 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H).
6,7-Dimethoxquinazolin-4(3H)-one10c: 4,5-Dimethoxy-anthranilic acid 9c gave 10c as brownish solid. Yield: 19%. M.p.290 °C–291 °C (EtOH) {lit.,
84
>260 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 7.96 (s, 1H), 7.42 (s, 1H), 7.11 (s, 1H), 3.88 (s, 3H), 3.85 (s, 3H).
Benzo[g]quinazolin-4(3H)-one10d: 3-Amino-2-naphthoic acid 9d gave 10d as a yellow solid. Yield: 61%. M.p. > 270 °C (EtOH) {lit.,
84
>260 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 12.05 (br. s, 1H), 8.82 (s, 1H), 8.45–8.16 (m, 2H), 8.12–8.02 (m, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H).
Pyrido[2,3-d]pyrimidin-4(3H)-one10e: 2-Amino nicotinic acid 9e gave 10e as a yellowish solid. Yield: 18%. M.p.256 °C–258 °C (EtOH) {lit.,
84
258 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 12.50 (br. s, 1H), 8.92 (dd, J = 4.5, 2.0 Hz, 1H), 8.47 (dd, J = 8.0, 2.0 Hz, 1H), 8.28 (s, 1H), 7.52 (dd, J = 8.0, 4.5 Hz, 1H).
2-Methylquinazolin-4(3H)-one10f: Using a modified literature procedure,79,80 a solution of anthranilic acid 9f (454 mg, 3.3 mmol) in acetic anhydride (2.0 mL) was heated at 120 °C for 3 h and the reaction mixture was evaporated to dryness. Ammonia solution (28% w/v; 4.0 mL) was added and the mixture was heated at reflux for 4 h and then left overnight at rt. The precipitate was collected by filtration, washed successively with water, then methanol and dried under reduced pressure, to give the quinazolinone 10f (200 mg, 38%) as white powder. M.p. 228 °C–230 °C {lit.,
80
230 °C–232 °C}. 1H NMR (300 MHz, (CD3)2SO): δ 12.14 (br. s, 1H), 8.05 (d, J = 7.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 2.20 (s, 3H).
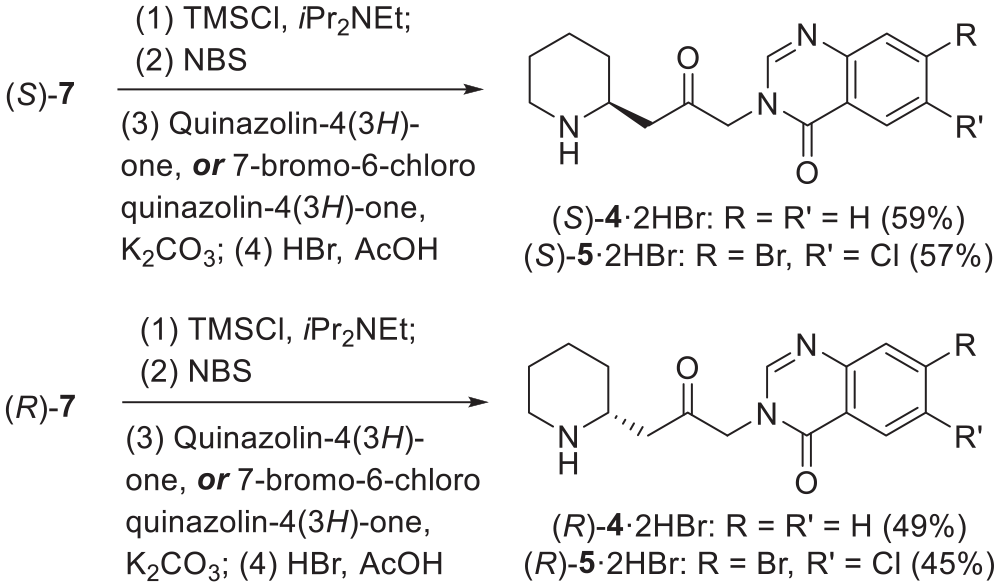
General procedure for synthesis of (–)-(2S)-2-[2-Oxo-3-(6-chloro-4-oxoquinazolin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12a: A mixture of commercially activated 4Å molecular sieves (0.5 g) and (−)-(S)-7 (300 mg, 1.09 mmol, 1.0 eq.) in dry CH2Cl2 (8 mL) was cooled to 0 °C, and left for 30 min. DIPEA (0.28mL, 1.61 mmol, 1.5 eq.) and TMSOTf (0.27 mL, 1.49 mmol, 1.4 eq.) were added, and the mixture was stirred at room temperature for 1 h. NBS (248 mg, 1.39 mmol, 1.3 eq.) was then added in one portion. The mixture was stirred at room temperature for 2h then poured into water (20 mL) and extracted with EtOAc (2 × 30 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (30 mL) and brine (30 mL), dried over anhydrous MgSO4, filtered and concentrated. A solution of the residue in dimethylformamide (DMF) (10 mL) was treated with 6-chloroquinazolin-4(3H)-one10a (273 mg, 1.51 mmol, 1.4 eq.) and K2CO3 (220 mg, 1.59 mmol, 1.5 eq.) and was stirred overnight at rt. The mixture was poured into H2O (60 mL) and extracted with EtOAc (2 × 60 mL). The combined organic layers were washed with H2O (60 mL) and brine (60 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford the crude product. This was purified by column chromatography (EtOAc), which afforded 12a as a white crystalline solid (58% over three steps). M.p.130 °C–133 °C; Rf = 0.45 (EtOAc); [α]D
20
= −12.0 (c = 1.00, acetone); HRMS (ES) calcd for [(C24H24N3O4
35
Cl + Na)]+476.1353, found 476.1335 (−3.8 ppm); IR: 3072, 2947, 1729, 1673, 1614, 1470, 1353, 1269, 1070, 850, 769, 733cm−1; 1H NMR (400 MHz, (CD3)2SO): δ 8.17 (s, 1H), 8.04 (d, J = 2.5 Hz, 1H), 7.86 (dd, J = 8.5, 2.5 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.32–7.24 (m, 5H), 5.06 (d, J = 12.5 Hz, 1H), 5.01 (d, J = 12.5 Hz, 1H), 4.96 (s, 2H), 4.72–4.64 (m, 1H), 3.87 (d, J = 14.0 Hz, 1H), 3.11 (dd, J = 16.0 Hz, 8.5 Hz, 1H), 2.88 (t, J = 12.5 Hz, 1H), 2.75 (dd, J = 16.0, 6.0 Hz, 1H), 1.63–1.47 (m, 5H), 1.34–1.21 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO) δ 202.2, 159.4, 154.9, 148.9, 147.1, 137.4, 135.1, 131.9, 130.0, 128.8, 128.2, 127.8, 125.5, 123.0, 66.6, 54.9, 47.2, 41.0, 40.0, 28.2, 25.4, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(6-Bromo-4-oxoquinazolin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12b: Following the general procedure outlined above 6-bromoquinazolin-4(3H)-one10b gave 12b as a white crystalline solid (53% overthree steps). M.p.128 °C–130 °C; Rf = 0.40 (EtOAc); [α]D
20
= –13.6 (c = 1.00, acetone); HRMS (ES) calcd for [(C24H24N3O4
79
Br+Na)]+520.0848, found 520.0856 (1.6 ppm); IR: 3069, 2944, 1729, 1674, 1614, 1468, 1353, 1269, 1073, 850, 769, 734cm-1; 1H NMR (400 MHz, (CD3)2SO): δ 8.18 (s, 1H), 8.17 (s, 1H), 7.97 (dd, J = 8.5, 2.5, 1H), 7.64 (d, J = 8.5 Hz, 1H), 7.33–7.22 (m, 5H), 5.06 (d, J = 12.5 Hz, 1H), 5.01 (d, J = 12.5 Hz, 1H), 4.96 (s, 2H), 4.72–4.65 (m, 1H), 3.87 (d, J = 13.0 Hz, 1H), 3.10 (dd, J = 16.0, 8.5 Hz, 1H), 2.87 (t, J = 12.0 Hz, 1H), 2.75 (dd, J = 16.0 Hz, 6.0 Hz, 1H), 1.63–1.46 (m, 5H), 1.33–1.18 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO)) δ 202.2, 159.3, 155.1, 149.0, 147.4, 137.9, 137.4, 130.1, 128.8, 128.6, 128.2, 127.8, 123.4, 120.1, 66.6, 54.9, 47.2, 41.0, 40.0, 28.2, 25.4, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(6,7-dimethoxy-4-oxoquinazolin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12c: Following the general procedure, 6,7-dimethoxyquinazolin-4(3H)-one10c gave 12c as a yellow solid (31% overthree steps). M.p. 96 °C–100 °C; Rf = 0.25 (EtOAc); [α]D
20
= –3.2 (c = 1.00, acetone); HRMS (ES) calcd for [(C26H29N3O6 + Na)]+502.1954, found 502.1973 (3.8 ppm); IR: 3059, 2921, 1667, 1607, 1498, 1370, 1271, 1018, 857, 782, 697cm-1; 1H NMR (400 MHz, (CD3)2SO): δ 8.03 (s, 1H), 7.4 (s, 1H), 7.35–7.24 (m, 5H), 7.13 (s, 1H), 5.06 (d, J = 12.5 Hz, 1H), 5.02 (d, J = 12.5 Hz, 1H), 4.91 (s, 2H), 4.72–4.65 (m, 1H), 3.88 (s, H, 3H), 3.87–3.84 (m, 1H), 3.83 (s, 3H), 3.11 (dd, J = 16.0, 8.5 Hz, 1H), 2.87 (t, J = 11.0 Hz, 1H), 2.72 (dd, J = 16.0 Hz, 5.8 Hz, 1H), 1.64–1.46 (m, 5H), 1.34–1.22 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 202.6, 159.7, 155.0, 149.2, 146.9, 144.6, 138.8, 137.4, 128.8, 128.2, 127.8, 114.9, 108.4, 105.5, 66.6, 56.4, 56.0, 54.8, 47.2, 41.0, 40.0, 28.2, 25.4, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(Benzo[g]-4-oxoquinazolin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12d: Following the general procedure, benzo[g]quinazolin-4(3H)-one10d gave 12d as a yellow solid (32% overthree steps). M.p.132 °C–134 °C; Rf = 0.40 (EtOAc); [α]D
20
= -2.2 (c = 1.00, acetone); HRMS (ES) calcd for [(C28H27N3O4+H)]+470.2080, found 470.2068 (–2.5 ppm); IR: 3060, 2929, 1721, 1667, 1618, 1428, 1355, 1268, 1070, 883, 742, 696cm-1; 1H NMR (400 MHz, (CD3)2SO): δ 8.83 (s, 1H), 8.26 (s, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.11–8.08 (m, 2H), 7.67 (t, J = 8.0 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.34–7.23 (m, 5H), 5.07 (d, J = 12.5 Hz, 1H), 5.02 (d, J = 12.5 Hz, 1H), 4.97 (s, 2H), 4.75–4.67 (m, 1H), 3.88 (d, J = 16.5 Hz, 1H), 3.13 (dd, J = 16.0 Hz, 8.5 Hz, 1H), 2.89 (t, J = 13.5 Hz, 1H), 2.76 (dd, J = 16.0 Hz, 6.0 Hz, 1H), 1.64–1.47 (m, 5H), 1.35–1.19 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 202.7, 160.9, 156.0, 147.6, 143.8, 137.4, 136.5, 131.5, 129.7, 129.1, 128.8, 128.3, 128.2, 128.0, 127.8, 127.0, 125.4, 120.7, 66.6, 54.7, 47.3, 41.0, 40.0, 28.2, 25.4, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(Pyrido[2,3-d]pyrimidin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12e: Following the general procedure, pyrido[2,3-d]pyrimidin-4(3H)-one10e gave 12e as a brown solid (30% overthree steps); Rf = 0.30 (EtOAc); [α]D
20
= –3.9 (c = 1.00, acetone); HRMS (ES) calcd for [(C23H24N4O4+H)] +421.1876, found 421.1892 (3.8 ppm); 1H NMR (400 MHz, (CD3)2SO): δ 8.97 (dd, J = 4.5, 2.0Hz, 1H), 8.50 (dd, J = 8.0, 2.0Hz, 1H), 8.36 (s, 1H), 7.57 (dd, J = 8.0, 4.5Hz, 1H), 7.35–7.24 (m, 5H), 5.07 (d, J = 12.5Hz, 1H), 5.02 (d, J = 12.5Hz, 1H), 4.98 (s, 2H), 4.70–4.64 (m, 1H), 3.87 (d, J = 14.5Hz, 1H), 3.12 (dd, J = 16.0Hz, 8.5Hz, 1H), 2.86 (t, J = 12.0Hz, 1H), 2.76 (dd, J = 16.0Hz, 5.5Hz, 1H), 1.63–1.45 (m, 5H), 1.33–1.18 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 202.2, 168.0, 160.0, 156.5, 151.5, 137.6, 137.4, 128.8, 128.2, 127.9, 123.3, 117.0, 66.6, 47.2, 41.0, 40.0, 31.1, 28.2, 25.4, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(2-methyl-4-oxoquinazolin-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12f: Following the general procedure, 2-methylquinazolin-4(3H)-one10f gave 12f as a white crystalline solid (50% overthree steps). M.p.128 °C–130 °C; Rf = 0.30 (EtOAc); [α]D
20
= –6.9 (c = 1.00, acetone); HRMS (ES) calcd for [(C25H28N3O4+H)]+434.2080, found 434.2097 (4.0 ppm); 1H NMR (400MHz, (CD3)2SO): δ 8.05 (d, J = 8.0Hz, 1H), 7.80 (t, J = 8.0Hz, 1H), 7.59 (d, J = 8.0Hz, 1H), 7.48 (t, J = 8.0Hz, 1H), 7.39–7.24 (m, 5H), 5.08 (s, 2H), 5.05 (s, 2H), 4.71–4.68 (m, 1H), 3.90 (d, J = 12.5Hz, 1H), 3.12 (dd, J = 16.0Hz, 8.0Hz, 1H), 2.95–2.80 (m, 2H), 2.50 (s, 3H), 1.67–1.49 (m, 5H), 1.35–1.23 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 203.0, 161.3, 155.3, 154.8, 147.5, 137.4, 135.0, 128.8, 128.2, 127.8, 127.0, 126.9, 126.6, 119.9, 66.7, 53.5, 47.4, 41.0, 40.0, 28.4, 25.4, 23.1, 18.7 ppm.
(–)-(2S)-2-[2-Oxo-3-(1,2,3-benzotriazin-4-oxo-4(3H)-yl)propyl]piperidine-1-carbamic acid benzyl ester 12g: Following the general procedure, 1,2,3-benzotriazin-4(3H)-one10g gave 12g as a white crystalline solid (26% over three steps). M.p.136 °C–138 °C; Rf = 0.75 (EtOAc); [α]D
20
= –3.0 (c = 0.5, acetone); HRMS (ES) calcd for [(C23H24N4O4+Na)]+443.1695, found 443.1680 (-3.4 ppm); IR: 3071, 2983, 1724, 1687, 1452, 1334, 1296, 1074, 889, 770, 748cm-1; 1H NMR (400 MHz, (CD3)2SO): δ 8.24 (d, J = 8.0 Hz, 2H), 8.11 (t, J = 8.0 Hz, 1H), 7.95 (t, J = 8.0 Hz, 1H), 7.38–7.23 (m, 5H), 5.39 (s, 2H), 5.12–5.01 (m, 2H), 4.77–4.68 (m, 1H), 3.89 (d, J = 16.5 Hz, 1H), 3.18 (dd, J = 16.5 Hz, 8.5 Hz, 1H), 2.78–2.95 (m, 2H), 1.44–1.68 (m, 5H), 1.37–1.20 (m, 1H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 202.8, 155.1, 154.8, 144.2, 137.4, 136.2, 133.7, 128.8, 128.6, 128.2, 127.9, 125.0, 119.6, 66.7, 58.5, 47.2, 41.0, 40.0, 28.3, 25.4, 18.7 ppm.
Deoxyfebrifugine analogues. General procedure: The Cbz-protected deoxyfebrifugine analogues (0.20 mmol) were dissolved in CH2Cl2 (1 mL) and cooled to 0 °C before 33% w/v HBr in acetic acid solution (1 mL) was added. The mixture was stirred at 0 °C for 15min before the cooling bath was removed and stirring was continued for 1h. Over this period rt was gradually reached. The mixture was diluted in methanol (5 mL), and the product was precipitated by addition of ether (30 mL). The supernatant was removed and this process was repeated three times. The product was dried under high vacuum to afford the desired salt.
(+)-Deoxychlorofuginonedihydrobromide13a: Following the procedure outlined above, 12a (50mg, 0.11 mmol) gave 13a·2HBr (45 mg, 85%) as a white solid powder. M.p.220 °C–222 °C (with dec.); HRMS (ESI) calcd for [(C16H20
35
ClN3O2–H)]+320.1166, found 320.1181 (4.7 ppm); IR: 3392, 2959, 2808, 2732, 1724, 1670, 1468, 1383, 1299, 1021, 839, 778, 639 cm-1; 1H NMR (400 MHz; (CD3)2SO): δ 8.56–8.40 (m, 2H), 8.30 (s, 1H), 8.05 (d, J = 2.5 Hz, 1H), 7.89 (dd, J = 8.5, 2.5 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 5.07 (d, J = 18.0 Hz, 1H), 5.00 (d, J = 18.0 Hz, 1H), 3.53–3.43 (m, 1H), 3.24 (d, J = 12.5 Hz, 1H), 3.02–2.87 (m, 3H), 1.80 (d, J = 10.0 Hz, 1H), 1.71 (app. d, J = 10.5 Hz, 2H), 1.64–1.38 (m, 3H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 201.2, 159.5, 148.9, 147.1, 135.3, 132.1, 130.1, 125.4, 123.0, 55.1, 51.9, 44.4, 42.9, 28.4, 22.2, 21.8 ppm.
(+)-Deoxybromofuginonedihydrobromide13b: Following the procedure outlined above, 12b (30 mg, 0.06 mmol) gave 13b·2HBr (25 mg, 80%) as a white solid powder. M.p.176 °C–178 °C (with dec.); HRMS (ES) calcd for [(C16H20
79
BrN3O2–H)]+364.0661, found 364.0678 (4.8 ppm); IR: 2925, 2720, 1711, 1658, 1492, 1386, 1294, 1073, 819, 695cm-1; 1H NMR (400 MHz; (CD3)2SO): δ 8.42 (br. s, 2H), 8.28 (s, 1H), 8.20 (d, J = 2.5 Hz, 1H), 8.01 (dd, J = 8.5, 2.5 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 5.06 (d, J = 18.0 Hz, 1H), 4.99 (d, J = 18.0 Hz, 1H), 3.53–3.43 (m, 1H), 3.24 (d, J = 11.5 Hz, 1H), 3.00–2.87 (m, 3H), 1.81 (d, J = 11.5 Hz, 1H), 1.71 (app. d, J = 11.5 Hz, 2H), 1.58–1.23 (m, 3H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 201.3, 159.4, 149.0, 147.4, 137.8, 130.2, 128.5, 123.3, 120.3, 55.1, 51.8, 44.4, 42.9, 28.5, 22.2, 21.7 ppm.
(+)-Deoxydimethoxyfebrifuginedihydrobromide13c: Following the procedure outlined above 12c (33 mg, 0.07 mmol) gave 13c·2HBr (22 mg, 62%) as a brownish solid powder. M.p.178 °C–180 °C (with dec.); HRMS (ES) calcd for [(C18H24N3O4)]+346.1767, found 346.1765 (-0.5 ppm); IR: 3358, 2942, 2799, 1698, 1609, 1452, 1385, 1287, 1193, 1002, 867, 767cm-1; 1H NMR (400 MHz; (CD3)2SO): 8.46 (br. s, 2H), 8.18 (s, 1H), 8.18 (s, 1H), 7.41 (s, 1H), 7.16 (s, 1H), 5.01 (d, J = 18.0 Hz, 1H), 4.95 (d, J = 18.0 Hz, 1H), 3.90 (s, 3H), 3.85 (s, 3H), 3.53–3.43 (m, 1H), 3.24 (d, J = 12.5 Hz, 1H), 3.02–2.87 (m, 3H), 1.81 (d, J = 15.5 Hz, 1H), 1.71 (app. d, J = 11.5 Hz, 2H), 1.62–1.36 (m, 3H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 201.6, 159.7, 155.2, 149.4, 147.0, 144.3, 114.8, 108.3, 105.4, 56.5, 56.3, 55.0, 51.9, 44.4, 42.9, 28.4, 22.2, 21.7 ppm.
(+)-Deoxynaphthafebrifuginedihydrobromide13d: Following the procedure outlined above 12d (70mg, 0.15 mmol) gave 13d·2HBr (62 mg, 83%) as a pale pink powder. M.p.229 °C–230 °C (with dec.); HRMS (ES) calcd for [(C20H23N3O2–H)]+336.1712, found 336.1697 (–4.5 ppm); 1H NMR (400MHz; (CD3)2SO): δ 8.84 (s, 1H), 8.43 (br. s, 2H), 8.28 (s, 1H), 8.22 (d, J = 5.5Hz, 2H), 8.12 (d, J = 8.5Hz, 1H), 7.68 (t, J = 7.5Hz, 1H), 7.59 (t, J = 7.5Hz, 1H), 5.06 (d, J = 18.0Hz, 1H), 4.98 (d, J = 18.0Hz, 1H), 3.54–3.44 (m, 1H), 3.23 (d, J = 12.5Hz, 1H), 3.03–2.88 (m, 3H), 1.82 (d, J = 12.0Hz, 1H), 1.71 (app. d, J = 10.0Hz, 2H), 1.60–1.36 (m, 3H) ppm; 13C NMR (100MHz, (CD3)2SO) δ 201.7, 161.0, 147.6, 143.6, 136.5, 131.6, 129.7, 128.3, 128.0, 127.12,125.354.9, 51.9, 44.4, 42.9, 28.5, 22.2, 21.8 ppm.
(+)-Deoxypyridinofebrifuginetrihydrobromide13e: Following the procedure outlined above 12e (100 mg, 0.24 mmol) gave 13e·3HBr (90 mg, 71%) as a yellowish solid powder. M.p. 264 °C–266 °C (with dec.); HRMS (ES) calcd for [(C15H18N4O2+H)]+287.1508 found 287.1512 (1.4 ppm); IR: 3026, 2931, 2779, 1723, 1693, 1614, 1421, 1374, 1336, 1217, 1161, 960, 881, 784, 724, 613cm-1; 1H NMR (400 MHz; (CD3)2SO): δ 9.01 (dd, J = 4.5, 2.0 Hz, 1H), 8.56 (dd, J = 8.0, 2.0 Hz, 1H), 8.50 (s, 1H), 8.46 (br. s, 2H), 7.62 (dd, J = 8.0, 4.5 Hz, 1H), 5.10 (d, J = 18.0 Hz, 1H), 5.03 (d, J = 18.0 Hz, 1H), 3.55–3.43 (m, 1H), 3.24 (d, J = 12.5 Hz, 1H), 3.01 (app. d, J = 6.5 Hz, 2H), 2.93 (dd, J = 10.0 Hz, 1H), 1.81 (d, J = 10.0 Hz, 1H), 1.71 (app. d, J = 10.0 Hz, 2H), 1.63–1.38 (m, 3H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 201.1, 160.8, 157.9, 156.2, 151.8, 136.9, 123.6, 117.2, 55.1, 51.9, 44.4, 42.9, 28.4, 22.2, 21.8 ppm.
(+)-Deoxymethylfebrifuginedihydrobromide13f: Following the procedure outlined above, 12f (80 mg, 0.18 mmol) gave 13f·2HBr (70 mg, 84%) as a whiteish solid powder. M.p.236 °C–238 °C (with dec.); HRMS (ES) calcd for [(C17H22N3O2+H)]+300.1707 found 300.1710 (1.09 ppm); IR: 3376, 2946, 2784, 1703, 1649, 1449, 1382, 1297, 988, 762, 695cm-1; 1H NMR (400 MHz, (CD3)2SO): 8.48 (br. s, 2H), 8.07 (d, J = 8.0 Hz, 1H), 7.85 (t, J = 8.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 5.18 (d, J = 18.5 Hz, 1H), 5.12 (d, J = 18.5 Hz, 1H), 3.54–3.44 (m, 1H), 3.24 (d, J = 13.0 Hz, 1H), 3.07 (app. dd, J = 6.0, 4.0 Hz, 2H), 2.99–2.87 (m, 1H), 2.45 (s, 3H), 1.80 (d, J = 9.0 Hz, 1H), 1.71 (app. d, J = 10.0 Hz, 2H), 1.61–1.40 (m, 3H) ppm; 13C NMR (100 MHz, (CD3)2SO): δ 201.7, 161.0, 156.3, 146.0, 135.5, 127.4, 126.8, 126.0, 119.6, 53.6, 51.8, 44.4, 43.1, 28.5, 23.0, 22.2, 21.8 ppm.