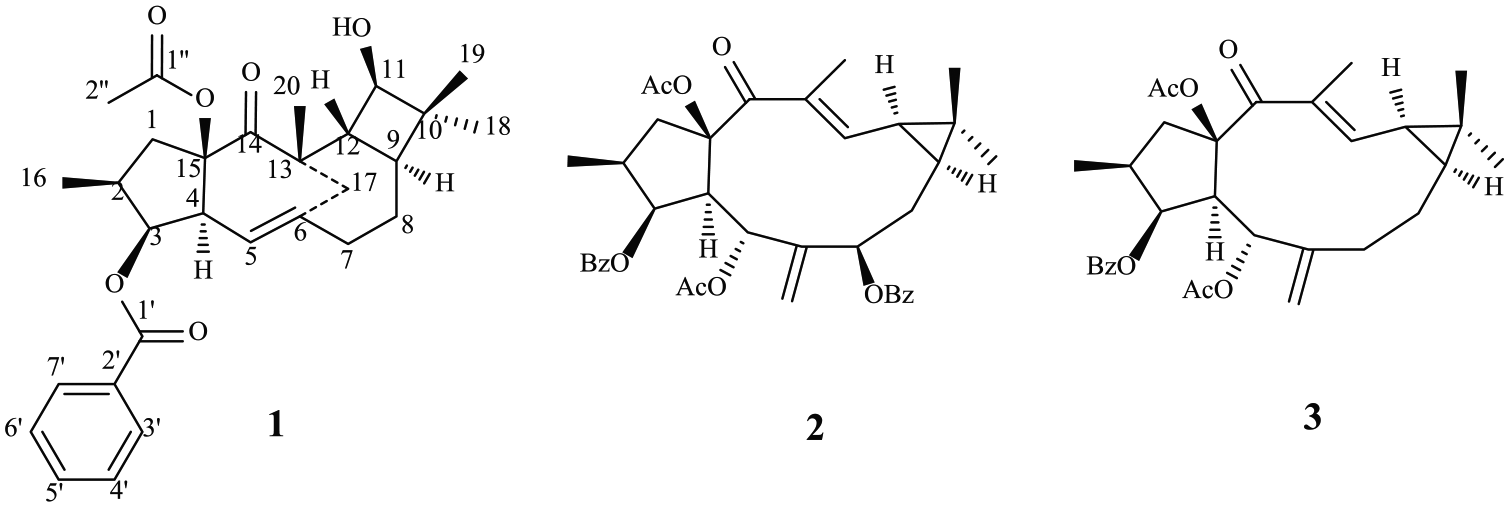
Eupholathone, a minor diterpenoid with an unusual tetracyclic skeleton, was obtained from the seeds of Euphorbia lathyris, along with two known lathyrane diterpenoids, euphorbia factors L2 and L3. The structure of eupholathone was elucidated by detailed interpretation of its spectroscopic data, especially two-dimensional nuclear magnetic resonance spectroscopy and high-resolution electrospray ionization mass spectrometry.
The Euphorbia genus, as the largest genus in the Euphorbiaceae family, comprises more than 2000 species throughout the world.1 The characteristic chemical constituents of the genus are diverse polycyclic diterpenoids, which attract considerable attention as their interesting bioactivities have potential for drug development.2–7 Ingenol-3-angelate (PEP005, ingenol mebutate), a cell death inducer and a protein kinase C (PKC) activator from Euphorbia peplus,8 has been approved by the Food and Drug Administration (FDA) for the treatment of actinic (or solar) keratosis, a disease stage associated with sun exposure which potentially can develop into skin cancer.9
Euphorbia lathyris, a well-known traditional Chinese medicine, is distributed throughout China. Its seeds have been widely used for centuries in China as a remedy for hydropsy, ascites, scabies, and snakebites.10 Previous studies on the chemical constituents of the plant resulted in the isolation of a series of diterpenoids, most of which were ingenane and lathyrane diterpenoids.11–22 Among these, the ingenane diterpenoids displayed PKC activating,23 and anticancer activities,24 whereas the lathyrane diterpenoids were demonstrated to show P-glycoprotein inhibitory,15,18 cytotoxic,20,21 and anti-inflammatory activities.22 In the present study, a new minor diterpenoid with an unusual tetracyclic skeleton, eupholathone (1), was isolated from the seeds of E. lathyris, together with two known lathyrane diterpenoids, that is, euphorbia factors L2 and L3 (Figure 1). The structure of 1 was determined by spectroscopic methods, including two-dimensional nuclear magnetic resonance spectroscopy (2D-NMR) (1H-1H COSY (correlation spectroscopy), HMQC (heteronuclear multiple quantum coherence), HMBC (heteronuclear multiple bond correlation), and NOESY (nuclear Overhauser effect spectroscopy)). Herein, the isolation and structural elucidation of these diterpenoids are reported.
Diterpenoids from Euphorbia lathyris.
Results and discussion
The molecular formula of 1 was assigned as C29H36O6 by the high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) peak at m/z 503.2399 ([M + Na]+; calcd 503.2404), and indicated the presence of 12 double-bond equivalents (DBEs). The infrared (IR) absorptions at 1734 and 1675 cm−1 indicated the presence of ketone and ester functionalities in 1. The 1H NMR spectrum of 1 (Table 1) displayed a doublet methyl at δ(H) 1.02 (3H, d, 8.8), an acetyl methyl at δ(H) 2.20 (3H, s), and three singlet methyls (δ(H) 0.86 (3H, s), 1.04 (3H, s), 1.19 (3H, s)). In addition, an olefinic proton at δ(H) 5.58 (1H, d, 5.3) and two oxygenated methines (δ(H) 5.72 (1H, t, 4.2) and 3.65 (1H, d, 8.6)) were observed in the 1H NMR spectrum. A mono-substituted phenyl (δ(H) 8.15 (2H, d, 7.3), 7.50 (2H, t, 7.7), and 7.63 (1H, t, 7.2)) could be easily distinguished in the 1H NMR spectrum. Consistent with the molecular formula of 1, 29 carbon signals comprising 8 quaternary carbons (3 sp3, 2 sp2, and 3 carbonyls), 12 CH (6 sp3 and 6 sp2), 4 CH2, and 5 methyls were observed in the 13C NMR spectrum. Since three carbonyls, a phenyl, and a double bond accounted for eight DBEs, the remaining four were attributed to the presence of a tetracyclic ring system in 1.
1H NMR and 13C NMR Data of 1 at 400 MHz in CDCl3.
Position
1H
13C
Position
1H
13C
1
3.63 (1H, m) 1.51 (1H, dd, 15.3, 8.3)
42.0
16
1.02 (3H, d, 8.8)
15.0
2
2.43 (1H, m)
37.6
17
2.01 (1H, d, 14.0) 2.19 (1H, d, 14.0)
45.6
3
5.72 (1H, t, 4.2)
79.2
18
0.86 (3H, s)
16.3
4
2.71 (1H, t, 4.8)
51.8
19
1.04 (3H, s)
28.5
5
5.58 (1H, d, 5.3)
119.1
20
1.19 (3H, s)
21.7
6
–
142.8
1′
–
166.2
7
2.41 (1H, m) 1.95 (1H, m)
37.4
2′
–
130.3
8
1.78 (1H, m) 1.23 (1H, m)
30.1
3′
8.15 (1H, d, 7.3)
129.7
9
1.48 (1H, m)
39.9
4′
7.50 (1H, t, 7.7)
128.5
10
–
38.8
5′
7.63 (1H, t, 7.2)
133.2
11
3.65 (1H, d, 8.6)
73.2
6′
7.50 (1H, t, 7.7)
128.5
12
2.47 (1H, m)
50.8
7′
8.15 (1H, d, 7.3)
129.7
13
–
51.2
1″
–
172.9
14
–
205.9
2″
2.20 (3H, s)
22.1
15
–
97.7
NMR: nuclear magnetic resonance.
Analysis of the 1H NMR, 13C NMR, and HMQC spectra of 1 enabled the assignment of all the protons to their bonding carbons. Three spin systems (a–c) drawn with bold bonds in Figure 2 could be plotted using the 1H-1H COSY spectrum assisted by the HMBC spectrum. The linkage of three structural fragments a–c by hetero-atoms and/or quaternary carbons was finally established by the HMBC experiment (Figure 2). The HMBCs of H2-17/C-5, H2-17/C-6, and H2-17/C-7 indicated the connectivity of two subunits a and b via C-6. The linkage of C-9 and C-11 via C-10 to form a cyclobutane in 1 was deduced by the HMBCs of H3-19/C-9, H3-19/C-10, and H3-19/C-11. The proton signal at δ(H) 2.71 (H-4) correlated with C-1, C-14, and C-15 in the HMBC spectrum, indicating the connectivity of C-1, C-4, and C-14 via C-15, which was further supported by HMBCs of H-1/C-4, H-1/C-14, and H-1/C-15. The linkage of C-20, C-12. and C-14 through C-13 was deduced by the HMBCs of H3-20/C-12, H3-20/C-13. and H3-20/C-14. The presence of the bridged ring (C-6–C-17–C-13) in 1 was evidenced by the HMBCs of H2-17/C-6, H2-17/C-12, and H2-17/C-13. The HMBC between H-3 and C-1′ showed that the benzoyloxy group was located at C-3. There was no observation of the HMBC between C-1″ and any proton signal except H3-2″, indicating that the acetoxyl group was anchored at the quaternary carbon C-15. Comparison of the chemical shift of C-15 in 1 with those of euphorbia factor L1116 and euphorbia factor L213 confirmed the above conclusion. The gross structure of 1 was thus outlined.
Selected 2D-NMR correlations of 1.
The relative configuration of 1 was deduced from the NOESY spectrum and coupling constant analysis. Comparison of the NMR data of the cylcopentane moiety in 1 with those of known diterpenoids also from this plant,13–16 especially the coupling constants of H-1, H-3, and H-4, indicated that the relative configuration of cylcopentane moiety in 1 was same as that of euphorbia factor L2. The conclusion presented here was confirmed by NOESY correlations of H-16/H-2″, H-2/H-4, and H-4/H-3. The NOESY correlations of H3-19/H-12 and H3-20/H-12 indicated that H-12, H3-19, and H3-20 were on the same side of the ring system and were tentatively assigned as the β-orientation. Therefore, the configuration of H3-18 was α. Also, the configuration of H-9 was assigned to be α by the NOESY correlation between H3-18 and H-9. Thus, the structure of eupholathone (1) was established as depicted.
The known diterpenoids were identified as euphorbia factor L2 (2)13 and euphorbia factor L3 (3)13 by comparison of their NMR, MS, and rotation data with those reported in the literature.
Experimental
TLC (thin layer chromatography): precoated SiO2 GF254 plates (Qingdao Haiyang Chemical Co., Ltd.); visualized by ultraviolet (UV) light (254 and/or 360 nm) and by spraying with 10% H2SO4 reagent followed by heating at 110 °C for 5 to 10 min. CC (column chromatography): silica gel (SiO2, 200–300 mesh; Qingdao Haiyang Chemical Co., Ltd.), and reversed-phase SiO2 (ODS-A 12 nm S-150; YMC Co.). All solvents were of analytical grade (Hangzhou Gaojing Fine Chemical Plant). Optical rotations: Rudolph-AutoPolIV polarimeter. IR spectra: Nicolet-Avatar-370 spectrometer (ν in cm−1). UV spectra: Shimadz-UV-2450 spectrometer. NMR spectra: Bruker AM-400 spectrometer (1H at 400 MHz, 13C NMR at 100 MHz, δ in ppm rel. to Me4Si, J in Hz). ESI-MS: Agilent-6210-LC/Tof mass spectrometer (in m/z).
Plant material was collected in the Xinjiang Uygur Autonomous Region of P.R. China, in October 2016, and identified as E. lathyris by Prof. Yong-Hong Zhang of the Fujian Medical University, P.R. China. A voucher specimen (No. ELS20161003) was deposited in the Zhejiang University of Technology.
The seeds of E. lathyris (3.0 kg) was pulverized and extracted with 95% EtOH exhaustively at room temperature to give a crude extract (465 g). The ethanol extract was solved in 3.0 L of CH3CN, and then extracted with cyclohexane for three times to remove fats. The CH3CN phase was evaporated under reduced pressure to afford a pale-yellow residue (58 g). The residue was then suspended in H2O (3.0 L), followed by CHCl3 extracting (5 × 0.5 L). The CHCl3 fraction (35 g) was applied to a silica gel CC eluted by petroleum/Me2CO (25:1 → 5:1) to give two main fractions, Frs.1–2. Compound 3 (6.5 g) was purified by recrystallization of fraction 1 (15.5 g) from petroleum ether/acetone (4:1). The filtrate was evaporated to give a white residue, which was further subjected to a reversed-phase silica gel column eluting with MeOH–H2O (5:5–7:3) yielded compounds 1 (3.0 mg). Fraction 2 (4.8 g) was separated by CC (SiO2, petroleum ether/acetone 15:1 → 10:1) to yield compound 2 (330 mg).
Eupholathone (1): white powder; m.p. 163–165 °C; [α]25D;78.0° (c 0.03, CHCl3); UV (CHCl3): λmax 270, 235 nm; IR (KBr): νmax 3378, 2956, 2911, 1734, 1675, 1450, 1375, 1180, and 1011 cm1; ESI-MS (pos.): 503 [M + Na]+; HR-ESI-MS for C29H36NaO6+ [M + Na]+ calcd 503.2404; found: 503.2399. 1H NMR and 13C NMR data: see Table 1.
Euphorbia factor L2 (2): white powder; m.p. 199–201 °C; [α]25D +100.8° (c 0.1, CHCl3); 1H and 13C NMR data were identical with literature data.13 ESI-MS (pos.): 665 [M + Na]+.
Euphorbia factor L3 (3): white powder; m.p. 152–153 °C; [α]25D +102.0° (c 0.1, CHCl3); 1H and 13C NMR data were identical with literature data.13 ESI-MS (pos.): 545 [M + Na]+.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support by the National Natural Science Foundation of China (81872777) is gratefully acknowledged.
ORCID iDs
Zha-Jun Zhan
Wei-Guang Shan
References
1.
JassbiAR.Phytochemistry2006; 67: 1977.
2.
ShiQWSuXHKiyotaH.Chem Rev2008; 108: 4295.
3.
VasasAHohmannJ.Chem Rev2014; 114: 8579.
4.
TianYGuoQLXuWD, et al. Org Lett2014; 16: 3950.
5.
WanLSShaoLDFuLB, et al. Org Lett2016; 18: 496.
6.
RawalMKShokoohiniaYChianeseG, et al. J Nat Prod2014; 77: 2700.
7.
LiuZGLiZLBaiJ, et al. J Nat Prod2014; 77: 792.
8.
HohmannJEvanicsFBertaL, et al. Planta Med2000; 66: 291.
9.
LebwohlMSwansonNAndersonLL, et al. Engl J Med2012; 366: 1010.
10.
ZhuAZhangTWangQ.J Ethnopharm2018; 277: 41.
11.
AdolfWHeckerEBalmainA, et al. Tetrahedron Lett1970; 11: 2241.
12.
ItokawaHIchiharaYYahagiM, et al. Phytochemistry1990; 29: 2025.
13.
AppendinoGTronGCCravottoG, et al. J Nat Prod1999; 62: 76.
14.
HohmannJEvanicsFVasasA, et al. J Nat Prod1999; 62: 176.
15.
AppendinoGPortaCDConseilG, et al. J Nat Prod2003; 66: 140.
16.
LiaoSGZhanZJYangSP, et al. Org Lett2005; 7: 1379.
17.
GaoSLiuHYWangYH, et al. Org Lett2007; 9: 3453.
18.
JiaoWDongWWLiZF, et al. Bioorg Med Chem2009; 17: 4786.
19.
LuJLiGYHuangJ, et al. Phytochemistry2014; 104: 79.
20.
LinMTangSZhangC, et al. Acta Pharm Sin B2017; 7: 59.
21.
WangJXWangQZhenYQ, et al. Chem Pharm Bull2018; 66: 674.
22.
ZhangCYWuYLZhangP, et al. J Nat Prod2019; 82: 756.
23.
HaslerCMAcsGBlumbergP.Cancer Res1992; 52: 202.
24.
KupchanSMUchidaIBranfmanAR, et al. Science1976; 191: 571.