Self-assembly of phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane,tetrasodium salt into a dimer through hydrogen bonding in water: Evidence from its encapsulation of monosubstituted benzenes,phenols,and polyaromatic hydrocarbons in water
Free accessResearch articleFirst published online January, 2020
Self-assembly of phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane,tetrasodium salt into a dimer through hydrogen bonding in water: Evidence from its encapsulation of monosubstituted benzenes,phenols,and polyaromatic hydrocarbons in water
Phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt, self-assembles into a dimer in water. The dimeric form was evidenced by its encapsulation of monosubstituted benzenes, phenols, and polyaromatic hydrocarbons. The stability constants of the inclusion complexes were determined by 1H nuclear magnetic resonance spectroscopy and the complexes formed were of 1:1 host to guest stoichiometry.
Self-assembled 3 encapsulates organic guest compounds in water.
Introduction
The construction of self-assembled molecules through non-covalent interactions has captured the attention of many research groups involved in molecular recognition studies during the last decade.1–3 Self-assembled molecules can be constructed through different kinds of non-covalent binding interactions, such as charge transfer, hydrogen bonding, metal co-ordination, and donor acceptor interactions.4–6 Of these interactions, hydrogen-bonded assemblies that possess a hydrophobic cavity for encapsulation of organic substrates are of great interest to us.
The encapsulation of organic substrates into the hydrophobic cavity of a host molecule offers an understanding of two important hydrophobic interactions, namely CH–π and π–π in host–guest chemistry.7,8 Recently, the behavior of hydrogen-bonded assemblies has been studied by many groups. However, to the best of our knowledge, we have reported the first water-soluble hydrogen-bonded hosts, which resulted from hydrogen bonding of carboxylic groups.9,10 In this article, we report another such host, a self-assembled dimer namely phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt (2). This dimer is formed through two hydrogen bonds involving phenolic groups, with evidence from its encapsulation of monosubstituted benzenes, phenols, and polyaromatic hydrocarbons in water.
Results and discussion
Phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt (2) (a light brown solid) was obtained in 20% yield by reacting chromotropic acid, disodium salt (1) with benzaldehyde in acidic medium under N2 at 90°C for 72 h. The structure was supported by the electrospray ionization (ESI) mass analysis, elemental analysis, and 1H and 13C nuclear magnetic resonance (NMR) spectroscopic data.
Dexanabinol, a dihydroxylated synthetic cannabinoid, was reported to self-assemble in water through the two hydroxy groups present in the molecule.11,12 The dimer of dexanabinol was found to be thermodynamically more favored than the individual dexanabinol or hydrates of dexanabinol in water. In addition, compound 1 was also reported to self-assemble in water through hydrogen bonding to form a tetramer.13
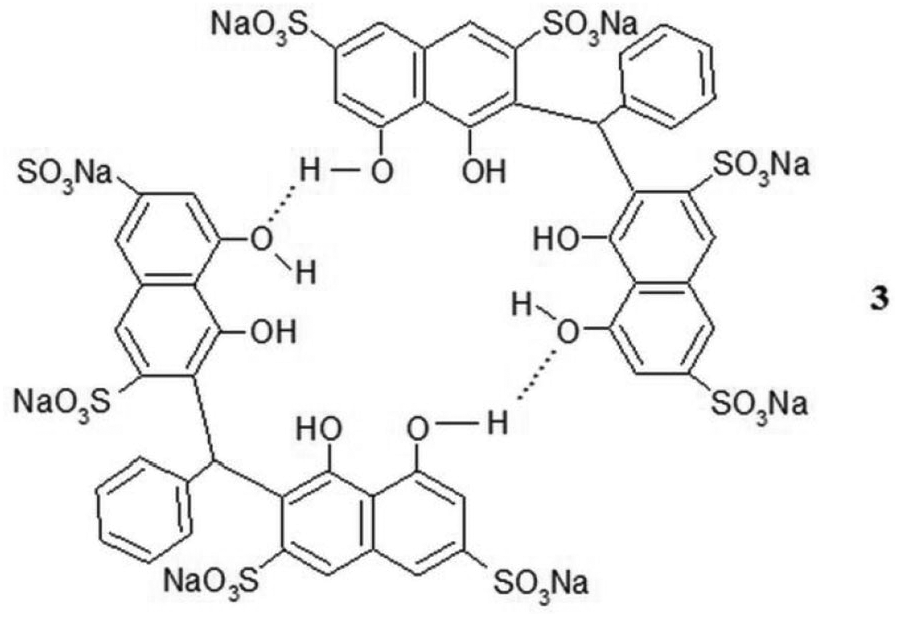
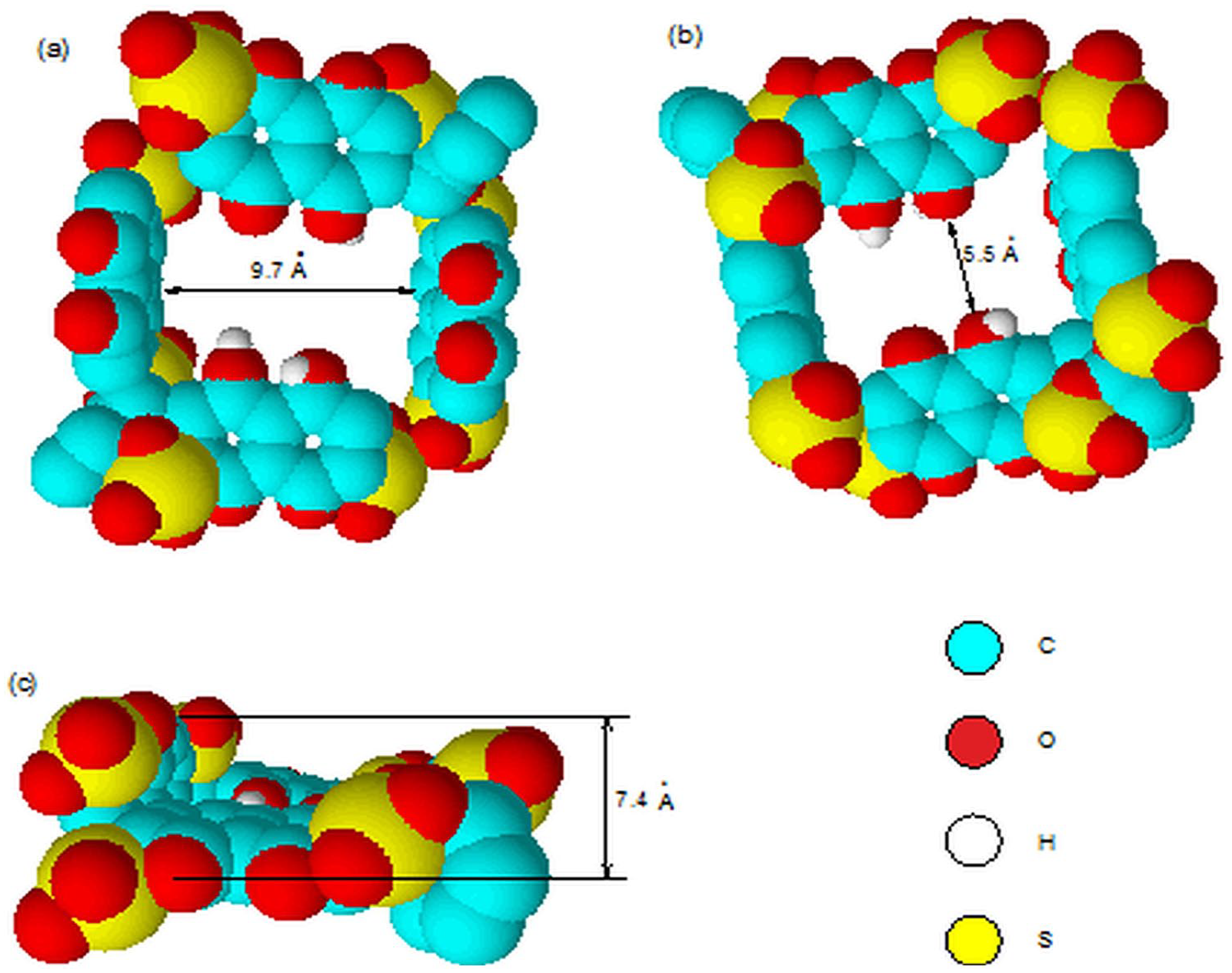
Since 2, which possesses two outer phenolic groups (the OH groups are not ionized, since the measured first pKa value is 7.5), is analogous to dexanabinol and compound 1, it is expected to form a dimer, being the more favorable species in the aqueous medium (through the two outer phenolic groups) than individual 2 or the hydrates of 2. Steric hindrance rules out the possibility of intermolecular hydrogen bonding involving the two inner phenolic groups. The self-assembly of 2 through hydrogen bonding was evidenced from a significant downfield shift of the naphthalene protons upon complexation with various guests. The appearance of sharp 1H NMR signals indirectly suggests that the hydrogen bonding does not lead to polymeric aggregates. Furthermore, the typical sharp singlet for the bridging methine group indicates the presence of benzaldehyde “bridge” between the two naphthalene rings restrict the rotation of 2, suggesting that the two naphthalene walls are perpendicular to each other instead of the broad signal for flexible host. The space-filling Corey–Pauling–Koltun (CPK) molecular model of the dimer of 2 (shown as 3) (Figure 1(a)–(c)) with a hydrophobic cavity resembles that of the cyclic tetramer, cyclotetrachromotropylene (4).14 It has two vertical naphthalene walls opposite to each other (shown as the right- and left-hand sides in Figure 1(a) and (b)) and the remaining two naphthalene walls are in a horizontal position (shown as the middle part of Figure 1(a)and (b)). Compound 2 is thus expected to act as a host molecule, since 4 was reported to encapsulate a variety of guests.14–17 This expectation is confirmed by the complexation study results of 2 with monosubstituted benzenes, phenols, and polyaromatic hydrocarbons as discussed in the following sections.
The CPK molecular models of 3 (the dimeric form of 2), forming an enclosed cyclic cavity: (a) Top view from the phenolic groups. (b) Top view from the sulfonic groups. (c) Side view.
Since this study is related to molecular recognition, it is necessary to ensure that the chemical shift changes of 2 are below the critical micelle concentration (CMC).18,19Figure 2 shows plots of the 1H NMR chemical shifts of 2 at various concentrations. The chemical shifts of the methine proton (H15) and benzyl ring protons of 2 (H12, H13, and H14; protons H13 and H14 are overlapped) stay constant (Δδ ⩽ 0.04 ppm, positive indicates downfield shifts) in the broad range of concentration (4.00 × 10−3 to 6.23 × 10−2 M) studied. On the contrary, the naphthalene ring protons of 2 (H6, H8, and H1) exhibited considerable downfield shifts (0.05, 0.06, and 0.07 ppm, respectively) when the concentration of 2 exceeded 1.80 × 10−2 M (log(2) > −1.74); however, there is no broadening of the NMR spectra. These small downfield shifts are an indication of hydrogen bond formation of 2 (hydrogen bonding decreases the electron density around the protons and thus moves the proton absorption to lower field) instead of aggregation (upfield shifts).19,20 The deshielding effect experienced by the naphthalene ring protons is slightly stronger compared to the benzyl ring protons which is due to the fact that the naphthalene units are involved in the hydrogen bonding. The further evidence for self-assemble of 2 came from the electrospray ionization mass spectrometry (ESI-MS) studies. In addition to the monomer peaks at m/z 820, there were significantly abundance of the dimer signals at m/z 1640 (100% intensity), compared to a minor aggregate of trimer at m/z 2420. This result suggests that 2 self-assembled predominantly as a dimer rather than other products. The analysis of 2 using high-performance liquid chromatography associated with diode-array detector in 0.1% acetic acid and acetonitrile (with gradient elution) at millimolar concentration displayed a single peak with a retention time of 3.259 min at multiple wavelengths (210, 254, and 276 nm, respectively), showing that 2 exists solely as a dimer in liquid solution. The formation of dimer for 2 in solution was further evidence from the infrared (IR) spectroscopy studies. It was noticed that there are two OH stretching vibrations occuring at 3440.94 and 3349.20 cm−1, respectively, suggesting that there are two OH groups. The free OH group is found at 3440.94 cm−1, while the OH with a hydrogen bond appeared at 3349.20 cm−1. The shift of OH group to a lower wave number is due to the fact that hydrogen bonding leads to a weakening of the OH group. In view of the fact that 2 is able to encapsulate monosubstituted benzenes, phenols, and polyaromatic hydrocarbons in water below the CMC, the plots in Figure 2 are better interpreted as 2 existing as dimeric 3, rather than in the monomeric form. Dexanabinol was also reported to be present as a dimer in water, even at very low concentration.11,12
1H NMR chemical shift (ppm) versus concentration of 2 in D2O at 25°C.
Complexation of monosubstituted benzenes
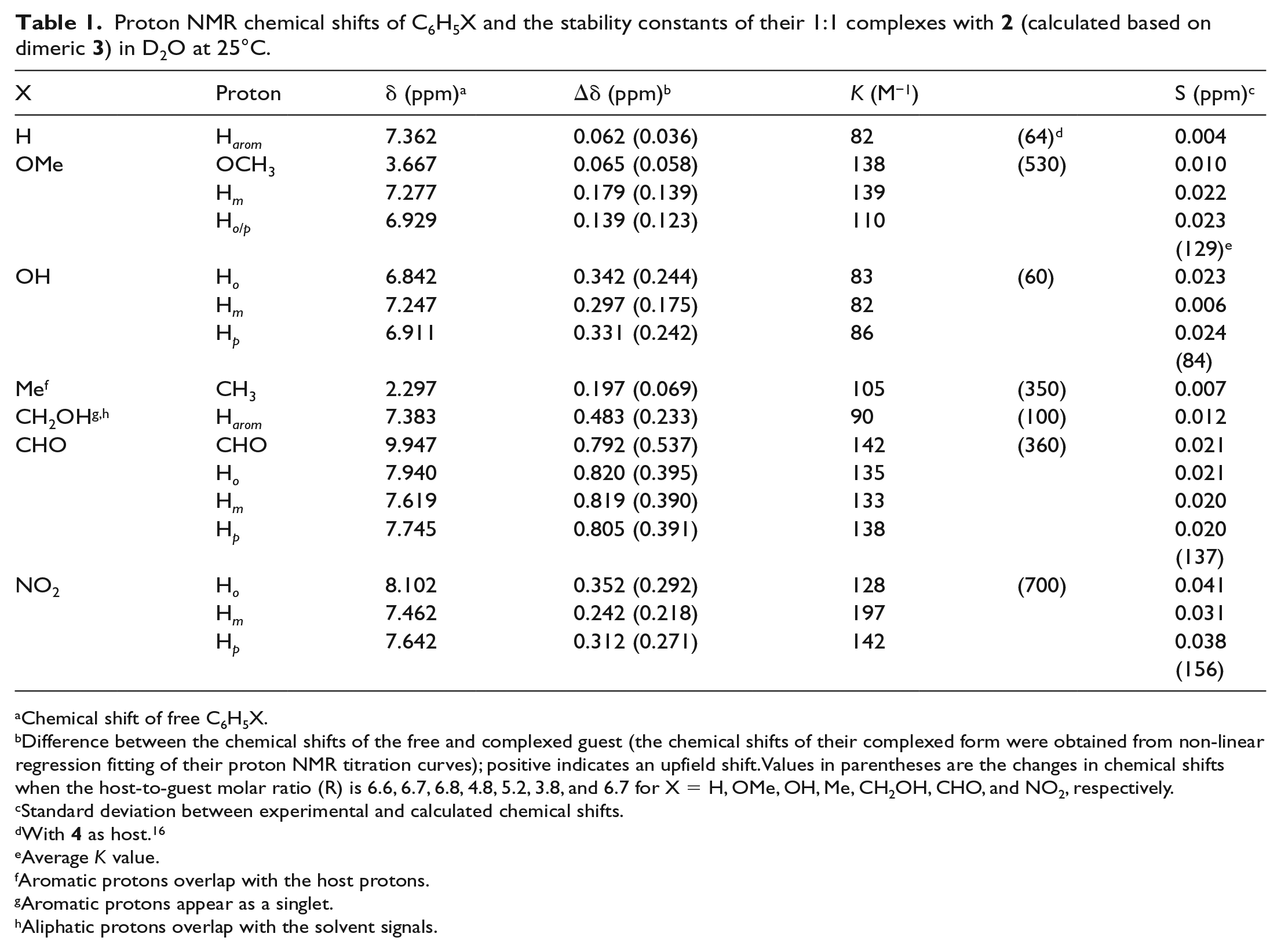
The self-assembly of 2 in water to form a dimer 3 with a hydrophobic cavity is best verified by its ability to encapsulate guest molecules. 1H NMR is a suitable method to use because the chemical shifts of the encapsulated molecule are expected to move to higher field due to the shielding effect of the ring current of the naphthalene walls enclosing the hydrophobic cavity. The encapsulation ability of 2 was first verified through a complexation study of 2 with seven monosubstituted benzenes (C6H5X, where X = H, OCH3, OH, CH3, CH2OH, CHO, and NO2) in D2O at 25°C using 1H NMR spectroscopy. These monosubstituted benzenes provided CH–π and π–π interactions with 2. It was observed that the proton chemical shifts of all the monosubstituted benzenes were shifted upfield in the presence of 2 (Table 1), indicating the presence of complexation. Figure 3 illustrates the change in the 1H NMR spectrum for nitrobenzene in the presence of 2.
Proton NMR chemical shifts of C6H5X and the stability constants of their 1:1 complexes with 2 (calculated based on dimeric 3) in D2O at 25°C.
Difference between the chemical shifts of the free and complexed guest (the chemical shifts of their complexed form were obtained from non-linear regression fitting of their proton NMR titration curves); positive indicates an upfield shift. Values in parentheses are the changes in chemical shifts when the host-to-guest molar ratio (R) is 6.6, 6.7, 6.8, 4.8, 5.2, 3.8, and 6.7 for X = H, OMe, OH, Me, CH2OH, CHO, and NO2, respectively.
Standard deviation between experimental and calculated chemical shifts.
Aliphatic protons overlap with the solvent signals.
1H NMR (400 MHz) spectra in D2O at 25 °C of 2.05 × 10−3 M nitrobenzene: (a) No host. (b) Is in the presence of 2 with the molar ratio of 2 (calculated based on dimeric 3) to nitrobenzene of 3.37.
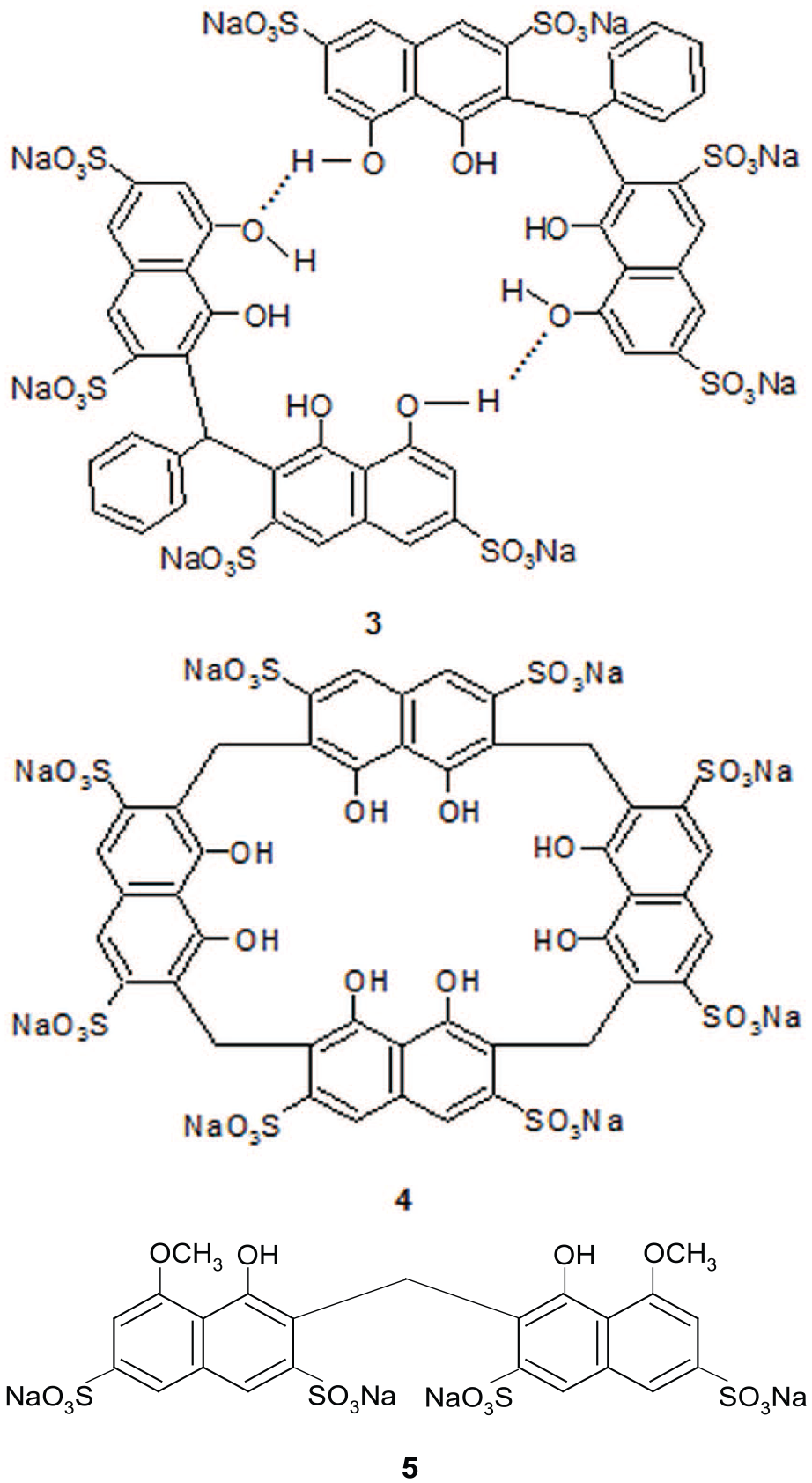
Using Job’s method,21–23 the stoichiometry for the formed complexes was found to be one molecule of 3 (that is, two molecules of 2) to one molecule of monosubstituted benzene, indicating a 1:1 host-to-guest stoichiometry. An example Job plot is shown in Figure 4 for nitrobenzene as the guest (plot based on 3 as host). From the plot, a maximum was obtained at the guest fraction of 0.5, indicating 2 is present in the dimeric form 3. This result is consistent with 2 being present in the dimeric form 3 enclosing a hydrophobic cavity to encapsulate the nitrobenzene guest. The self-assembly of 2 through hydrogen bonding of the two outer phenolic OH groups is further supported by the inability of compound 5 (Figure 5) to complex guest molecules in water.24 The absence of outer phenolic groups in 5 prevents the formation of intermolecular hydrogen bonding and thus no dimer is formed and hence no hydrophobic cavity.
Job plot of the complex formed between 3 with guest nitrobenzene against the mole ratio of guest.
The dimeric structure 3, cyclic tetramer 4 and non-dimeric 5.
The almost equal changes in the chemical shifts of the aromatic protons of each monosubstituted benzene (Table 1) indicate that the aromatic guest molecule lies horizontally in the host cavity of 3. Taking benzaldehyde as an example, the Δδ values for CHO, Ho, Hm, and Hp are, respectively, 0.792, 0.820, 0.819, and 0.805 ppm. These results indicate that the benzaldehyde guest molecule lies horizontally in the host cavity of 3.
Similar to the case of 4 as host, the variation in the values of the stability constant K, which was obtained through a non-linear regression fitting procedure (a typical fitting curve is shown in Figure 6 for benzaldehyde as guest)25 can be attributed to two factors, namely the electronic effect of X on the benzene ring and the interaction between the C–H bond, if any, of X and the π—bonds of the host.16 For example, nitrobenzene gives a more stable complex compared to benzene (K values respectively of 156 and 82 M−1) because the electron-withdrawing nitro group decreases the electron density of the aromatic ring, resulting in a better π–π interaction with the electron-rich naphthalene walls of the host. If the electronic effect, which is caused by the substituent X, is the only factor that contributes to the K values of the complexes formed, then toluene and anisole are expected to give K values smaller than that of benzene due to poorer π–π interactions, since their substituents, CH3 and OCH3, are electron donors. However, their K values are larger (105 and 129 M−1, respectively) because of the three C–H bonds (from CH3 and OCH3), which may provide CH–π interactions with the host. On average, the K values are only about three times smaller than the corresponding K values with 4 as the host (Table 1). These results are consistent with 3 having a bigger hydrophobic cavity than 4.
Proton chemical shift titration curves of the aldehyde proton of benzaldehyde (1.8 × 10−3 M) in D2O at 25 °C. [H]o/[G]o is the molar ratio of host 3 to the benzaldehyde guest.
Complexation of phenols
The complexation of six phenolic guests (p-cresol, p-nitrophenol, p-bromophenol, p-methoxyphenol, sodium p-sulfonatophenol, and phenol) with 2 was also studied in D2O at 25°C using 1H NMR spectroscopy. These guests provided di-substitution effects and π–π interactions with the host. The proton chemical shifts of all the phenols are shifted upfield (Table 2) in the presence of 2. Figure 7 shows the change in the 1H NMR spectrum of p-cresol in the presence of 2. Job plots indicate the complexes are of 1:1 host (3) to guest stoichiometry (an example is given in Figure 8 for p-nitrophenol as the guest, plot based on 3 as the host). These results again support the presence of dimeric 3 in water.
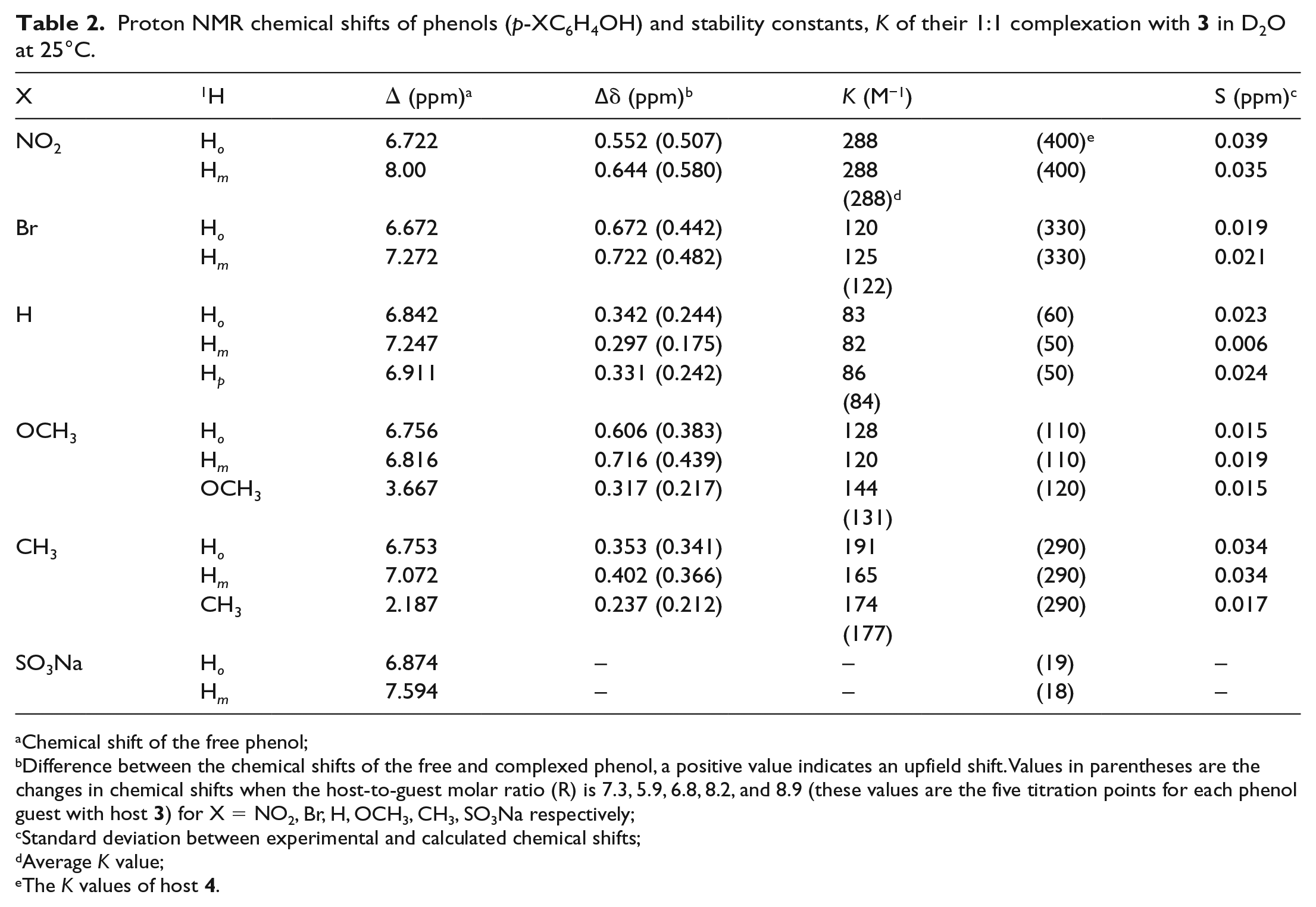
Proton NMR chemical shifts of phenols (p-XC6H4OH) and stability constants, K of their 1:1 complexation with 3 in D2O at 25°C.
Difference between the chemical shifts of the free and complexed phenol, a positive value indicates an upfield shift. Values in parentheses are the changes in chemical shifts when the host-to-guest molar ratio (R) is 7.3, 5.9, 6.8, 8.2, and 8.9 (these values are the five titration points for each phenol guest with host 3) for X = NO2, Br, H, OCH3, CH3, SO3Na respectively;
Standard deviation between experimental and calculated chemical shifts;
Average K value;
The K values of host 4.
1H NMR (400 MHz) spectra in D2O at 25 °C of 2 × 10−3 M p-cresol: (a) p-cresol in the absence of 2. (b) p-cresol in the presence of 2 with the molar ratio of 3 to p-cresol of 4.49.
Job plot of the complex formed, [HG], between 3 and the guest p-nitrophenol against the mole ratio of guest.
There are three possible modes of penetration of phenols into the cavity of 3 as shown in Figure 9(a)–(c). They are (a) penetration from the substituent X (NO2, Br, H, CH3, and OCH3), (b) penetration from the hydroxy end, and (c) horinzontal penetration. If the penetration is from the substituent X, then the chemical shift changes (∆δ) of Hm (in relation to OH) should be larger than those of Ho. Taking p-nitrophenol and p-methoxyphenol as examples, the ∆δ values of Hm is slightly higher than the Ho protons, but the differences are quite small, 0.09 ppm (0.645–0.552 ppm) and 0.11 ppm (0.716–0.606 ppm), respectively (Table 2), regardless of whether the substituents are electron-withdrawing groups (e.g. NO2) or electron-donating groups (e.g. CH3O). If the penetration is from (b), the hydroxy end, then the ∆δ values of Ho should be larger than Hm. However, the higher ∆δ values of Hm than Ho in most of the cases (taking p-bromophenol as an example, Hm = 0.722 and Ho = 0.672 ppm, respectively), rules out this possibility. In addition, the ∆δ values of Hm and Ho protons of p-bromophenol, phenol, and p-cresol are practically the same (∆δ of about 0.05 ppm or below), indicating that the more favorable inclusion of the phenols is by either horinzontal penetration or from shallow penetration from the X-substituent end as shown in (c). It is interesting to note that for 4, which has a smaller cavity, the phenols penetrated from the more hydrophobic substituent X, instead of OH.16 These results are again consistent with 3, having a bigger hydrophobic cavity than 4.
Three possible modes of inclusion of p-cresol in the cavity of 3: (a) Inclusion from the substituent end. (b) Inclusion from the hydroxy end. (c) Horizontal inclusion.
In the case of p-sodium sulfonatophenol, there is no change in the proton chemical shifts in the presence of 3, indicating weak or no complexation. The possible reasons that may account for weak complexation are (1) the unfavorable electrostatic interaction between the SO3− groups of both the guest and host, and (2) the SO3− group of the guest is better solvated by water molecules rather than in the hydrophobic cavity of the host.
The stability constant, K, calculated using a non-linear regression fitting procedure (a typical plot is shown in Figure 10 for p-methoxyphenol as guest) varied between 80 and 300 M−1, regardless of whether the p-substituted groups were electron-withdrawing (NO2, 288 M−1; Br, 123 M−1) or electron donors (CH3, 177 M−1; OCH3, 131 M−1), or phenol itself (84 M−1). These small differences in the K values indicate that the electronic effect of the substituent is not as important when compared to the π–π or CH–π interactions between the phenols and the naphthalene walls of the host. On average, the K values are about 1.5 times smaller when compared to 4 as the host (Table 2).
Variation of proton chemical shifts of p-methoxyphenol (1.50 × 10−3 M) with the molar ratio of host 3 to guest, R, in D2O at 25 °C.
Complexation of polyaromatic hydrocarbons
A third piece of evidence consistent with 2 existing in the dimeric form 3 in water came from our study on the complexation of four polyaromatic hydrocarbons (naphthalene, fluorene, phenanthrene, and chrysene). The low solubility of polyaromatic hydrocarbons in water rules out the use of the 1H NMR technique. A simple and convenient method to study the complexation of polyaromatic hydrocarbons in an aqueous medium is the transport method.15 The rate of transporting a polyaromatic hydrocarbon from one hexane phase to another through an aqueous phase in a U-tube will be increased in the presence of 2 in the aqueous phase if it exists as dimeric 3 that can encapsulate the polyaromatic hydrocarbon.
The transport rates of all the four hydrocarbons indeed increase when 2 is present in the aqueous phase, further supporting the presence of dimeric 3 in the aqueous medium. A typical absorbance (of the polyaromatic hydrocarbon transported) versus time plot is shown in Figure 11 for phenanthrene as the guest.
Transport of phenanthrene through water at 29°C: (a) Without host and (b) In the presence of host 3 with a concentration of 1.701 × 10−3 M.
Since the complexes monosubstituted benzenes and phenols are of 1:1 host (3) to guest stoichiometry, we assume the same stoichiometry for the polyaromatic hydrocarbon complexes to calculate the stability constant values (Table 3), using the equations reported earlier.15 The K values of these bigger guest molecules are large and comparable to those reported by the cyclic host 4, and they increase with the number of aromatic rings. A plot of log K versus the number of aromatic rings9,10 (Figure 12) yields a linear line with a slope of 0.69 (correlation coefficient of 0.970), giving an aromatic π–π interaction energy of −0.95 Kcal/mol (slope multiplied by −2.303 RT (where R is the gas constant and T is the absolute temperature).9 This value (0.69) is in agreement with the results reported for other aromatic hosts9,10 and is close to the value of −1 Kcal/mol calculated by Burley and Petsko from the interaction between two phenyl rings in proteins.26
Stability constants K of complexes formed between polyaromatic hydrocarbons and 3 in water.
Relationship between log K and the number of aromatic rings (N) in the aromatic hydrocarbon guests. The numbers refer to the number of aromatic rings of the aromatic hydrocarbons as given in Table 3.
Ionization of the phenolic groups would destroy the dimeric structure of 3. We repeated the transport experiment of chrysene in the presence of sodium hydroxide in the aqueous phase (0.001 M of 3 in the presence of 0.001 M of sodium hydroxide). As expected, the transport rate was reduced by 60%, indicating a reduction of concentration of self-assembled 3. This revealed that the intermolecular hydrogen bonding was formed in 2.
Conclusion
Phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt (2) self-assembles into a dimer through hydrogen bonding in water as evidenced from its encapsulation with monosubstituted benzenes, phenols, and polyaromatic hydrocarbons in water. The complexation is of 1:1 host (dimer 3) to guest stoichiometry.
Experimental
Materials used in this study are as follows: chromotropic acid, disodium salt, benzaldehyde, hydrochloric acid, sodium hydroxide, monosubstituted benzenes, phenols, and polyaromatic hydrocarbons were commercial samples.
Synthesis of phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt (2)
The product 2 was synthesized by refluxing chromotropic acid, disodium salt (0.5 g), (dissolved in 4 mL of H2O) with distilled benzaldehyde (1 mL) and concentrated HCl (8 mL, 37%) at 90°C under N2 for 72 h. The product 2 was neutralized to pH 5 with 1 M sodium hydroxide solution. The resulting solution (about 150 mL) was concentrated until approximately 15 mL of solution remained. It was then allowed to precipitate at room temperature. The precipitate was filtered and dried at 60°C–70 °C on a hot plate producing a crude brown solid. The brown solid was further purified by column chromatography using sephadex G-15 hydrophilic as a separation medium and water as the eluent. The light brown product 2 gave an Rf value of 0.83 (silica gel thin-layer chromatography, eluent: n-BuOH/EtOH/H2O (1:1:1). Yield of 2 was 20%, mp > 260 °C.
Analytical calculated for (C27H16S4O16Na4).11 H2O: C, 31.95; H, 3.75; Na, 9.07. Analytical found: C, 32.19; H, 3.68; Na, 9.25. 1H NMR (D2O at 25 °C, solvent peak at 4.70 ppm as internal reference): δ = 8.44 (2H, s, H1), 8.00 and 7.96 (2H, d, H8, J 2.0 Hz), 7.35 and 7.34 (2H, d, H6, J 2.0 Hz), 7.24 and 7.06 (5H, multiplet, H12, H13 & H14), 7.28 (1H, s, methane, H15). 13C NMR (D2O at 25 °C, TMS as external reference): δ 37.7 = methine carbon (C15) and 14 aromatic carbons at 109.4 (C6), 117.1 (C9), 118.9 (C8), 120.5 (C3), 126.1 (C1), 127.1 (C12), 128.3 (C13), 128.6 (C14), 133.7 (C10), 139.9 (C2), 141.8 (C7), 143.7 (C11), 149.8 (C4), and 152.2 (C5). The assignment of 1H and 13C chemical shifts was accomplished after analyzing the DEPT, HMQC, and HMBC spectra. ESI (MeOH): peak for monomer at m/z 820 [M + 4NaH], dimer at m/z 1640 [M + 4NaH + 3MeOH] and trimer at m/z 2420 [M + 4NaH + 3MeOH] with relative abundance of 5.0:5.0:1.1, respectively.
13C NMR (75 MHz) spectra were recorded with a Bruker Avance 300 NMR spectrometer and 1H NMR (400 MHz) spectra with a Bruker Avance 400 NMR spectrometer. The solvent peak at 4.70 ppm (unaffected by the concentration variation of the host and guest compounds) was used as the internal reference for all the 1H NMR spectra. In all the 1H NMR chemical shift titrations, the concentration of monosubstituted benzenes and phenols was kept constant at about 2 × 10−3 M, while the concentration of 2 varied.
Job method of continuous variations was performed according to the reported procedure.21–23 Different proportions of equal concentrations of 2 (calculated based on dimeric 3) and the guests were mixed (total volume of the mixture kept constant) and their 1H NMR spectra recorded.
Transport experiments using the U-tubes were carried out at room temperature (27 °C–29 °C) as described previously.15 The concentrations of the polyaromatic hydrocarbons in the hexane source phase varied from 7.5 × 10−4 to 6.0 × 10−2 M, while that of 2 (calculated based on dimeric 3) in the aqueous phase was 5 × 10−3 M.
The pKa determination of 2 was carried out by measuring the ultraviolet (UV) spectra of 2 in buffers ranging from pH 5 to 13, as described in the literature.27 From the UV spectra, the first and second pKa values obtained were 7.5 and 11.5, respectively.
Calculations of the stability constants K of the 1:l host to guest complexes were performed using the non-linear regression fitting of the proton chemical shift titration curves for the phenols and monosubstituted benzenes25 and using the equations for the transport experiment for polyaromatic hydrocarbons.15 They have an estimated error of 10%.
Mass spectra were recorded using a Finnigan TSQ 7000 mass spectrometer and fast atom bombardment (FAB) spectra were obtained using a Finnigan MAT95XL-T mass spectrometer at the National University of Singapore.
Fourier-transform infrared (FTIR) spectra were measured by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) (Perkin Elmer Spectrum 100). The spectra were obtained in the wavenumber range between 4000 and 650 cm−1 in 12 scans at resolution of 0.5 cm−1 and rationed against a background spectrum.
High-performance liquid chromatography was performed using a HPLC unit (Agilent Technologies, California, US) that consists of a Agilent 1260 Infinity Quaternary Pump, a 1260 Infinity Diode-Array Detector, a Agilent 1260 infinity standard autosampler injector with a loop of 10 µL, and a reversed phase Agilent ZORBAX Eclipse XDB 5 µm C18 column (150 mm × 4.6 mm). Gradient elution of two solvents was used: Solvent A consisted of 0.1% acetic acid in water and solvent B was HPLC-grade acetonitrile. The gradient elution was programmed as follows: 35% A initially, 40% A at 3 min, 25% A at 10 min, 15% A at 14 min, and 10% A at 15 min. An additional of 5 min was included to reach the initial conditions and to achieve mobile phase stabilization. The flow rate was 0.8 mL min−1, temperature was set at 25 °C and qualitative analyses were carried out at 210, 254, and 276 nm, respectively.
Supplemental Material
supplimental_material – Supplemental material for Self-assembly of phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt into a dimer through hydrogen bonding in water: Evidence from its encapsulation of monosubstituted benzenes, phenols, and polyaromatic hydrocarbons in water
Supplemental material, supplimental_material for Self-assembly of phenyl-bis-(4,5-dihydroxy-2,7-disulfonato-3-naphthyl)methane, tetrasodium salt into a dimer through hydrogen bonding in water: Evidence from its encapsulation of monosubstituted benzenes, phenols, and polyaromatic hydrocarbons in water by Chen Son Yue and Bo Long Poh in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank the Malaysian Government for providing an FRGS grant to carry out this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Malaysian Government by an FRGS grant (no. 304.PKIMIA.670002).
ORCID iD
Chen Son Yue
Supplemental material
Supplemental material for this article is available online. Additional information of characterization of compound 2: 1H, 13C, DEPT90, HMQC and HMBC NMR spectra; ESi-Q mass spectrum; HPLC chromatogram and IR spectrum.
References
1.
BeckerRReckGRadegliaR, et al. J Mol Struct2006; 784: 157–161.
2.
LiXGongSLYangWP, et al. Tetrahedron2008; 64: 6230–6237.
3.
MackovaMHimlMBudkaJ, et al. Tetrahedron2013; 69: 1397–1402.
4.
LehnJM. Supramolecular chemistry: concepts and perspectives. Weinheim: VCH Wiley, 1995, pp. 1–10.
5.
WeiYGuoXShuangS, et al. J Photoch Photobio B2005; 81: 190–194.
MoralesASantanaAAlthoffG, et al. J Organomet Chem2011; 696: 2519–2527.
23.
ItoSKogameCAkashiM, et al. Tetrahedron Lett2016; 57: 5243–5245.
24.
TeemCM. Synthesis and complexation study of a derivatized cyclotetrachromotropylene in an aqueous solution. Gelugor, Malaysia: Universiti Sains Malaysia, 2004, pp. 44–51.
25.
PohBLTeemCM. Tetrahedron2005; 61: 5123–5129.
26.
BurleySKPetskoGA. Science1985; 229: 23–29.
27.
PohBLLimCS. Tetrahedron1990; 46: 3651–3658.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.