
Abstract
A mild, efficient, and transition-metal-free catalytic strategy is developed to construct thioesters via selective N–C bond cleavage of Boc2-activated primary amides. This strategy is successfully carried out with stoichiometric Na2CO3 as the base and provides the corresponding products in moderate to excellent yields.

Introduction
The thioester group is a fundamental structural motif which is found widely in polymers, agrochemicals, pharmaceuticals, and natural products (Figure 1).1–7 This class of compound is usually employed as key intermediates to synthesize some important skeletal units, such as β-lactams, 8 β-lactones 9 esters,10,11 aldehydes, 12 and ketones.13,14 Thioesters also play important roles in several biological processes.3,15,16 During the past decades, significant research efforts have focused on the synthesis of thioesters. The condensation reaction between carboxylic acid derivatives and thiols was the initial synthetic strategy toward thioesters (Scheme (1a)).15,17–20 In 1997, Xiao and Alper 21 reported the first palladium-catalyzed method for the synthesis of thioesters via carbonylation of aryl iodides and n-BuSNa. Since then, transition-metal-catalyzed thiocarbonylation processes have attracted much attention and various methods have been explored to construct thioester compounds using aryl halides and S-nucleophiles (Scheme (1b)).22–31 Moreover, oxidative coupling between aldehydes and thiols (or disulfides) has also offered a reliable solution to obtain thioester compounds (Scheme (1c)).32,33 However, in principle, metal catalysts or stoichiometric oxidants were needed in the above reaction processes. In view of environmental concerns, metal-free and oxidant-free synthetic methods are highly desirable for the preparation of thioesters.
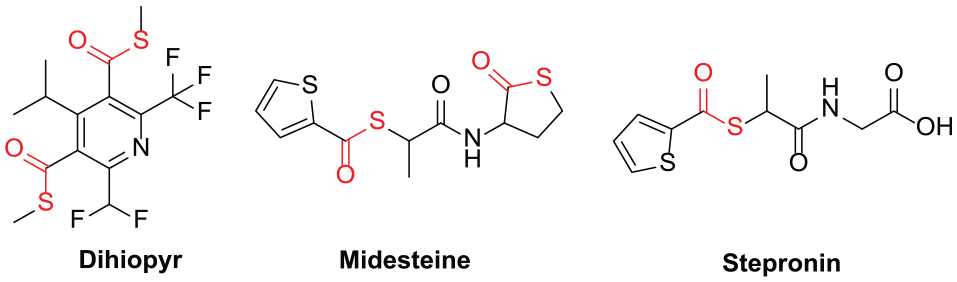
Examples of pharmaceuticals containing a thioester motif.
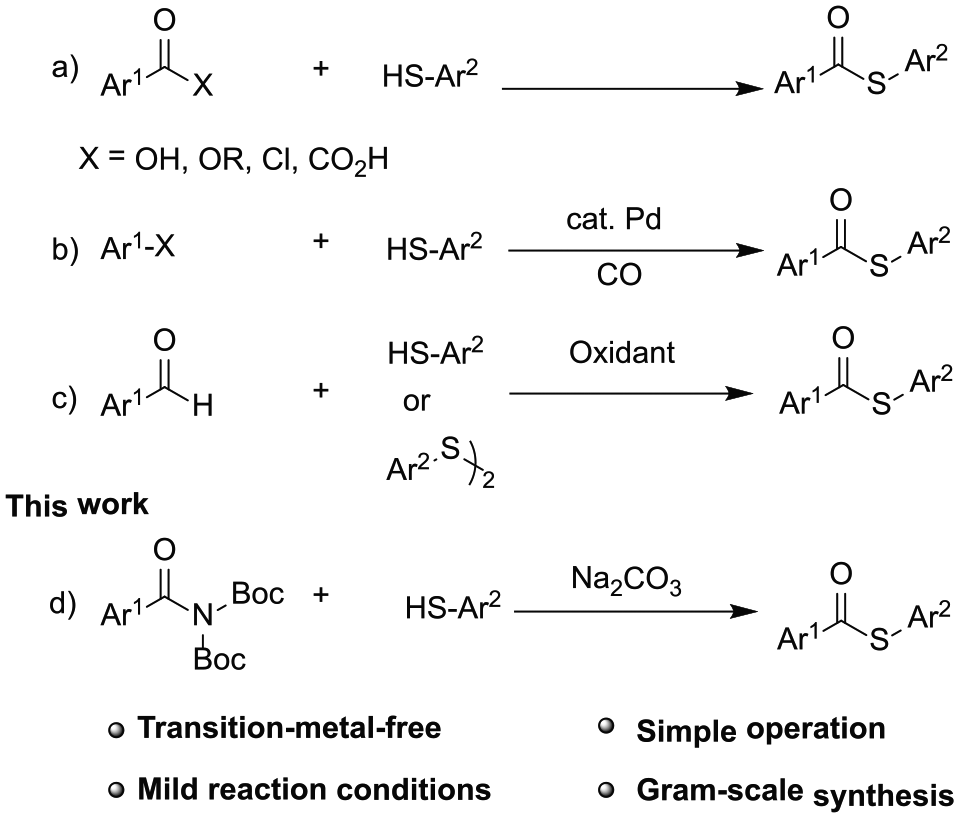
Synthesis of thioesters.
In recent years, significant breakthroughs have been exploited to construct C–C or C–X bonds via transition-metal-catalyzed amide N–C bond cleavage.34–38 In 2015, Hie et al. 39 reported the first nickel-catalyzed conversion of amides into esters via selective N–C bond cleavage. Soon after, Suzuki,40–48 Negishi,49–51 borylation,52,53 Heck,53,54–60 Sonogashlira, 61 and other cross-coupling reactions54,52,62–73 have been extended by this means.
However, the geometries of typical amide bonds are planar as a result of amidic resonance, which results in amides having very stable chemical bonds (15–20 kcal mol−1). 40 Many studies have shown that distortion of amide bonds greatly affects the stability and reactivity of amides. In recent work, cross-coupling of twisted amides with arenes, 67 amines,74–77 alcohols,48,58 and phenols 78 have been successfully explored and the twisted amide bond is considered as a controlling factor in selective amide bond activation.79–81 Considering the reactivity of twisted amides, herein, we report a synthetic strategy for the preparation of thioesters using twisted amides and thiols without a transition-metal-catalyst.
Results and discussion
Initially, we carried out the reaction with N,N-di-Boc-activated amide82–85
Optimization of the Reaction Conditions. a


DABCO: 1,4-diazabicyclo[2.2.2]octane; DBU: 1,8-diazabicyclo[5.4.0] undec-7-ene; THF: tetrahydrofuran; DCE: 1,2-dichoroethane; DMF: dimethylformamide; N.D.: not detected.
Reaction conditions:
Isolated yield.
Base (0.5 equiv.).
Base (1.0 equiv.).
rt.
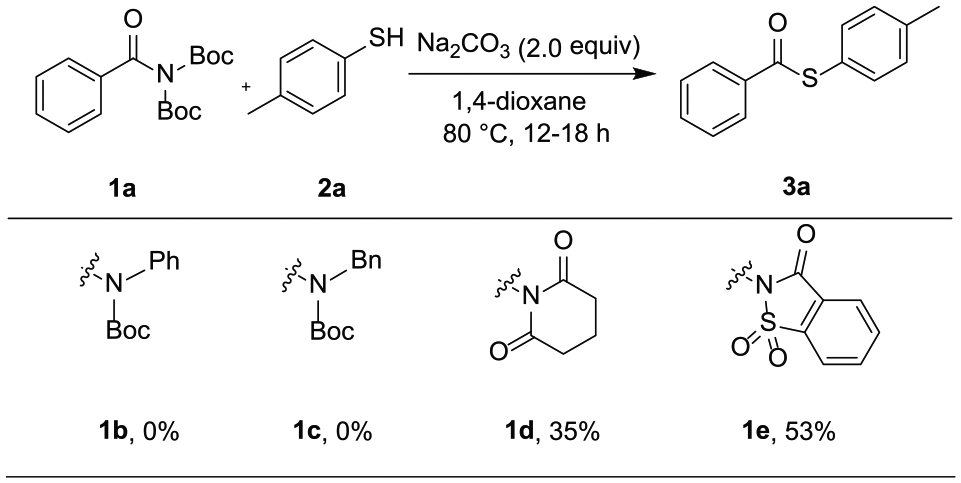
Evaluating different amides.
With optimized reaction conditions in hand, the substrate scope of the amides was evaluated. As shown in Scheme 3, the reactions of electron-rich amides were well-tolerated and afforded the corresponding thioesters

Substrate scope of amides.
Next, we tested the tolerance of thiols under the optimized conditions (Scheme 4). Both electron-donating and electron-withdrawing substituents on the phenyl afforded the desired products

Substrate scope of thiols.

One-pot experiment.
To check the practicality of this transformation, a gram-scale reaction (10 mmol) was carried out (Scheme 6). The reaction afforded product

Scale-up experiment.
Conclusion
In summary, we have reported a transition-metal-free cross-coupling reaction for the preparation thioester compounds using commercial primary amides and thiols via activated N–C bond cleavage under exceedingly mild conditions. The transformation is accomplished efficiently with the assistance of stoichiometric Na2CO3 under an air atmosphere. A high-yielding gram-scale reaction demonstrated the potential value of the synthetic utility of this method.
General procedure
All reagents and solvents were commercially available and used without further purification. Unless otherwise noted, all reactions were run under a nitrogen atmosphere. Purification of all products was carried out by flash chromatography using brand 200–300 mesh silica gel. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Ascend instrument at 400, 100 and 376 MHz, respectively. Chemical shifts were reported in δ (ppm) referenced to an internal tetramethylsilane (TMS) standard for 1H NMR (δ 0.00), and CDCl3 (δ 77.16) for 13C NMR. The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, hept = heptaplet, m = multiplet, and br = broad. High-resolution mass spectra (HRMS) were obtained on an Agilent mass spectrometer using electrospray ionization-time of flight (ESI-TOF). GC-MS spectra were recorded on a Shimdazu-GC-MS 2010QP-Ultra instrument.
Preparation of the starting materials
General procedure for N,N-Boc2-amide synthesis
A previously published procedure was followed. An oven-dried round-bottomed flask (100 mL) equipped with a stir bar was charged with the primary amide (8.26 mmol, 1.0 equiv.), 4-dimethylaminopyridine (DMAP) (typically, 0.10 equiv.), and dichloromethane (typically, 25 mL), placed under a positive pressure of nitrogen, and subjected to three evacuation/backfilling cycles under high vacuum.
84
Di-tert-butyl dicarbonate (typically, 2.0 equiv.) was added portion-wise to the reaction mixture with vigorous stirring at 0 °C, and the reaction mixture was stirred overnight at room temperature. After the indicated time, the reaction mixture was concentrated and unless stated otherwise, purified directly by chromatography on silica gel (hexanes/ethyl acetate) to give analytically pure product.
General procedure for synthesis of N-Boc amides from secondary amides (1b , 1c )
An oven-dried round-bottomed flask (100 mL) was charged with a secondary amide substrate (5.0 mmol, 1.0 equiv.), DMAP (0.1 equiv.), and dichloromethane (typically, 0.20 M).
86
Di-tert-butyl dicarbonate (1.0 equiv.) was added in one portion, and the reaction mixture was allowed to stir at room temperature for 15 h. After the indicated time, the reaction mixture was quenched with NaHCO3 (aq, 10 mL), extracted with EtOAc (3 × 20 mL), washed with H2O (1 × 20 mL), and brine (1 × 20 mL). The organic layer was dried, and concentrated. Unless stated otherwise, purification by flash chromatography (EtOAc/hexanes) afforded the pure product. In our hands, the N-Boc activation of secondary amides typically proceeds in average yields of >80%.
General procedure for N-acyl-glutarimide synthesis (1d )
An oven-dried round-bottomed flask (100 mL) equipped with a stir bar was charged with amine (8.84 mmol, 1.0 equiv.), triethylamine (typically, 2.0 equiv.), DMAP (typically, 0.25 equiv.), and dichloromethane (typically, 50 mL), placed under a positive pressure of nitrogen, and subjected to three evacuation/backfilling cycles under high vacuum.
87
Acyl chloride (typically, 1.1 equiv.) was added dropwise to the reaction mixture with vigorous stirring at 0 °C, and the reaction mixture was stirred overnight at room temperature. After the indicated time, the reaction mixture was diluted with Et2O (20 mL) and filtered. The organic layer was washed with HCl (1.0 N, 30 mL) and brine (30 mL), dried, and concentrated. Unless stated otherwise, the crude product was purified by recrystallization (toluene) to give analytically pure product.
General procedure for saccharinamide synthesis (1e )
A previously published procedure was followed.
88
An oven-dried flask (25 mL) equipped with a stir bar was charged with the amine (typically, 3.0 mmol, 1.0 equiv.), triethylamine (typically, 1.0 equiv.), and N,N-dimethylacetamide (DMAc, typically, 0.75 M), placed under a positive pressure of nitrogen, and subjected to three evacuation/backfilling cycles under high vacuum. The acyl chloride (typically, 1.0 equiv.) was added dropwise to the reaction mixture with vigorous stirring at 0 °C, and the reaction mixture was stirred for 1 h at room temperature. After the indicated time, the reaction mixture was diluted with H2O (5 mL). The resulting solid was collected by filtration, washed with Et2O (1 × 10 mL), and dried. The crude product was purified by recrystallization (methanol or toluene) to give analytically pure product.
General procedure for the synthesis of S-p-tolyl benzothioate (e.g. 3a )
A mixture of
One-pot experiment
To a dry flask was added primary amide
S-(p-tolyl) benzothioate (
S-(p-tolyl) 4-methylbenzothioate (
S-(p-tolyl) 4-methoxybenzothioate (
S-(p-tolyl) 4-fluorobenzothioate (
S-(p-tolyl) 4-chlorobenzothioate (
S-(p-tolyl) 4-bromobenzothioate (
S-(p-tolyl) 4-(trifluoromethyl)benzothioate (
S-(p-tolyl) pyridine-3-carbothioate (
S-(p-tolyl) furan-2-carbothioate (
S-(p-tolyl) naphthalene-2-carbothioate (
S-(4-methoxyphenyl) benzothioate (
S-(4-(tert-butyl)phenyl) benzothioate (
S-(o-tolyl) benzothioate (
S-(4-fluorophenyl) benzothioate (
S-(4-chlorophenyl) benzothioate (
S-(4-bromophenyl) benzothioate (
S-(4-(trifluoromethyl)phenyl) benzothioate (
S-(3,4-dimethylphenyl) benzothioate (
S-(perfluorophenyl) benzothioate (
S-(naphthalen-2-yl) benzothioate (
S-cyclopentyl benzothioate (
S-cyclohexyl benzothioate (
S-benzyl benzothioate (
S-phenethyl benzothioate (
Supplemental Material
supplementary_material – Supplemental material for Na2CO3-promoted thioesterification via N–C bond cleavage of amides to construct thioester derivatives
Supplemental material, supplementary_material for Na2CO3-promoted thioesterification via N–C bond cleavage of amides to construct thioester derivatives by Jiasi Tao, Weijie Yu, Jin Luo, Tao Wang, Wanling Ge, Ziwei Zhang, Bingjie Yang and Fei Xiong in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this paper.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this paper: We gratefully acknowledge the NSFC (21562026, 21762025) and the Natural Science Foundation of Jiangxi Province (20161BAB203085) for financial support.
Supplemental material
Supplemental material for this paper is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.