
Abstract
Charge transport rate is one of the key parameters determining the performance of organic electronic devices. Based on density functional theory, exchange-correlation functionals which adequately account for non-covalent interactions, such as M06-2X and wB97XD, should significantly improve the accuracy of charge transport rate calculations for large systems with non-covalent interactions. In this work, the B3LYP hybrid functional, the variant hybrid functional M06-2X, and the long-range-corrected wB97XD functional were used to perform geometry optimizations and charge transport rate calculations on 11 variants of tetrabenzo[a,d,j,m]coronene, including tetrabenzo[a,d,j,m]coronene itself and its tetra-substituted and octa-substituted derivatives. Our results indicate that the molecular geometries of these benzocoronene semiconductors are large quasi-planar conjugated π systems, and the incorporation of different substituents significantly affects their frontier molecular orbitals. The hole carrier mobility (µ+) and electron carrier mobility (µ−) of the methoxy-substituted derivatives (
Keywords
The charge transport rate of tetrabenzo[a,d,j,m]coronene derivatives was calculated using density functional theory method with three different functionals, respectively.

Introduction
Semiconductors are light, flexible, easy to chemically modify, and able to form large films at low temperatures. Thus, research on the use of cheap organic semiconductors on microelectronic and optoelectronic devices has recently received increasing levels of attention. 1 Since carrier transport is an extremely important physical process in semiconductors, charge transport rate is one of the key parameters determining the performance of organic electronic devices. In the aggregated state, the structure, molecular orientation, and condensed phase formed by molecular aggregation of a semiconductor are important factors affecting its charge transport properties. To design novel organic semiconductors that will improve the performance of modern devices, it is necessary to understand the relationship between an organic semiconductor’s charge transport properties and its structure. At present, highly reliable and computationally cheap quantum chemical methods, such as density functional theory (DFT), are being widely used in the field of organic optoelectronics to validate the results of experimental studies and to predict the performance of novel organic semiconductor molecules. Based on DFT quantum chemistry, researchers have calculated and predicted the intrinsic electron and hole transport rates of organic molecular systems with various molecular structures, substituents, or hybridizations, including molecular derivatives such as thiophene, 2 bithiazole, 3 tetracene, 4 anthracene,5,6 pentacene,7–9 heptacenes, 10 diindole–diimide, 11 perylene,12,13 triphenylene,14,15 truxene,16,17 coronene, 18 and phthalocyanine.19,20 These theoretical studies on the relationship between the molecular structures of different systems and their charge transport properties are extremely useful for guiding experimental studies.
Aromatic compounds with large conjugated π-electron systems are an important class of materials for organic semiconductors. In particular, discotic liquid crystals (DLCs), composed of a rigid core surrounded by flexible alkyl chains, are a novel type of aromatic organic semiconductor.21,22 DLCs have excellent solubility in common solvents and can self-assemble in a temperature-dependent manner to form highly ordered columnar phases. These molecules are also capable of self-healing, and their charge carriers can migrate between the aromatic cores of the columns. DLCs are therefore a typical one-dimensional semiconductor material. Benzocoronene derivatives are a class of DLCs with high intrinsic carrier mobility. In particular, hexa-peri-hexabenzocoronenes (
Electron energy band models and discontinuous hopping models are often used to simulate charge transport between adjacent semiconductor molecules. Energy band models are usually suitable for inorganic semiconductors with periodic structures at low temperatures.31,32 Hopping models, on the contrary, provide reasonable predictions for the charge mobility trends of organic semiconductors that exhibit significant temperature dependencies at room temperature.33–35 A number of studies have used discontinuous hopping models to simulate and calculate carrier mobility during charge transport processes. In theory, if the exact exchange-correlation function is known, DFT can produce extremely accurate quantum chemistry calculations. Significant efforts are being made to develop accurate exchange-correlation functionals for property calculations using a range of DFT functionals, from local density functionals to doubly hybridized functionals.30,36–38 In the literature, the B3LYP hybridized functional, which contains 20% Hartree–Fock (HF) exchange, is frequently used to calculate the carrier mobility of charge transport processes. 39 The aromatic compounds of the conjugated π-electron system aggregated into a one-dimensional semiconductor via non-covalent interactions, such as π–π interactions, Van der Waals forces, hydrogen bonds, halogen bonds, or CH–π interactions. DFT methods such as M06-2X, CAM-B3LYP, LC-ωPBE, and wB97XD, which adequately account for non-covalent interactions, should be better suited, compared with B3LYP, for calculating the charge mobility of large conjugated π-electron systems that aggregate via non-covalent interactions. Our group has previously tested the variant M06-2X hybrid functional for carrying out quantum calculations on the charge mobility of benzocoronene semiconductor molecules. 18 Furthermore, we have evaluated the use of B3LYP, M06-2X, and long-range-corrected CAM-B3LYP and wB97XD functionals for calculating the charge mobility of hybrid phthalocyanine–tetrabenzoporphyrin macrocycles. 19
In this work, the B3LYP, M06-2X, and wB97XD functionals were used with the 6-31+G(d,p) basis set to perform geometry optimizations and charge transport rate calculations on 11 tetra- and octa-substituted
We studied how different numbers and types of substituents in the rigid aromatic core of tetrabenzocoronenes affected their charge transport properties. These substituents include –F, –Cl, –CH3, –OCH3, and –CN.
We evaluated the strengths and weaknesses of density functionals when calculating the carrier mobility of large conjugated π-electron systems, including functionals such as M06-2X, wB97XD, and B3LYP.
Theoretical methodology
All calculations were performed using the Gaussian 09 software package 40 and the B3LYP hybrid functional; 41 the variant hybrid functional M06-2X, 42 HF exchange integral component of which is 56%; and long range correction functional wB97XD43,44 of DFT, 45 HF exchange integral component of which are short-range (α) 22.2%, long-range (β) 100%, ω = 0.2. The different functions and 6-31+G(d,p) basis set were first used to optimize the geometries of the ground state for neutral molecules and charged counterparts. The frequency calculations were performed at the same level based on the optimized geometries. All the frequencies are real, showing that all the geometries obtained are energy minima on the potential energy surfaces. Subsequently, we had calculated the energy of the highest occupied molecular orbital (HOMO) and the LUMO, the HOMO–LUMO energy gap of the single-molecule optimized structure, the internal reorganization energies for hole and electron transfer (λ+, and λ−, respectively), and the charge transfer matrix element for hole and electron transfer (t+, and t−, respectively) of the dimer.
The mobility of the charge carrier μ can be obtained using the Einstein equation, which is expressed as
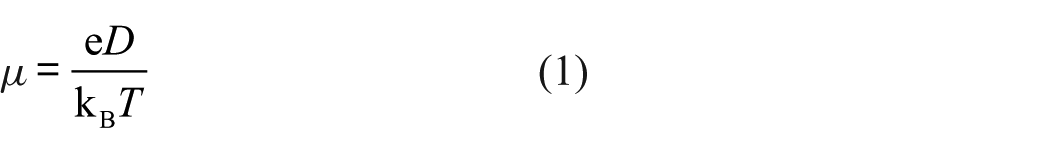
where e is the electron charge (1.60 × 10−19 C), kB is the Boltzmann constant (1.38 × 10−23 J/K), T is the absolute temperature, and D is the average diffusion coefficient of the charge starting from a molecule and toward all directions in three-dimensional space, as given by equation (2). The diffusion coefficient is expressed as

where ri is the distance between adjacent molecules, ki is the charge transfer rate constant between adjacent molecules, and pi is the probability, denoted by

of charge transfer to molecule i.
The π-conjugated organic semiconductor materials have the properties of one-dimensional charge carrier migration. The average diffusion coefficient can be simplified as

where r is the disk spacing between adjacent discotic molecules, and k is the charge transfer rate constant between adjacent molecules. The carrier mobility is obtained by substituting into equation (1)

According to the semi-classical model of Marcus charge transfer,46–50 the constant of charge transfer rate between adjacent molecules is expressed as follows

where h is the Planck constant (6.626 × 10−34 J s), t is the charge transfer matrix element, λ is the charge reorganization energy, and T is the absolute temperature. Under a certain temperature, λ and t are the main parameters affecting the charge transport rate constant. To achieve a larger charge transport rate constant, the molecule should have smaller reorganization energy λ and larger intermolecular charge transport matrix element t.
The reorganization energy λ was directly calculated using the insulation potential surfaces, that is, the recombination energy λ+ of the transmission hole and the recombination energy λ− of the transmission electrons are calculated as follows


where E(Ar+/Ar) represents the total energy of the cationic single point energy based on the Ar neutral molecular configuration optimization, and E(Ar+/Ar+) represents the total energy when the Ar+ ion is optimized configuration.
The charge transfer matrix element characterizes the coupling strength of electron–electron interaction, and several methods have been proposed to evaluate the transfer integral within a molecular dimer. The simplest way is the frontier orbital energy level splitting method.51,52 That is, the closed shell system was formed by adding one electron to the molecular/molecular cation system. The energy level splitting of HOMO and HOMO – 1 at the transition state was calculated. One half is the hole transfer matrix element t+. The closed shell system is formed by removing one electron from the molecular/molecular anion system. The energy level splitting of LUMO and LUMO + 1 at the transition state is calculated. One half is the negative charge transfer matrix element t−. The transfer matrix elements for holes and electrons can be estimated by halving these values. The mean squares of the charge transfer matrix elements of the coronene derivatives were calculated using equation (7) according to the Boltzmann distribution using the energies of the dimers at different dihedral angles (Ei) and the corresponding matrix elements (ti) at a temperature of 298.15 K. The root of <t2> is the charge transfer matrix element t. 53 The carrier transport rates, µ, were also calculated for the nine molecules using equations (3) and (4)

Results and discussion
Molecule design and geometry optimization
Geometry optimizations and frequency calculations were performed on the 11 molecules shown in Figure 1 (

Molecular structures of tetrabenzo[a,d,j,m]coronene and its derivatives.
Structure-optimized dihedral angles (θ in degree) a for coronene derivatives using different methods.

θ1 = ∠1-2-3-4, θ2 = ∠1-2-5-6, θ3 = ∠4-3-2-5 and θ4 = ∠3-2-5-6.
The angles footnote references Figure 2 where the atoms are defined.

Optimized structures of coronene derivatives
It is worth noting that single-crystal experiments have shown that tetramethyl-substituted derivative
Frontier molecular orbitals
Frontier molecular orbitals are an important factor for determining the electrical properties of materials. The energy levels of a material’s HOMO and LUMO directly affect the effective injection capability between a pair of electrodes in practical applications. The intrinsic frontier orbital energies of the 11 molecules were calculated at the B3LYP/6-31+G(d,p) level of theory. The energy gap of organic semiconductors ranges roughly between 1.4 and 4.2 eV (135.08–405.24 kJ/mol). In Figure 3, it is shown that the energy gap of

HOMO energies (EH), LUMO energies (EL), and HOMO–LUMO gaps (Eg) of tetrabenzo[a,d,j,m]coronene and its derivatives at the B3LYP/6-31+G(d,p) level.
Figure 3 illustrates the HOMO and LUMO frontier orbitals and the energy gap diagrams of
Reorganization energy of the molecules
The hole and electron transport reorganization energies of the 11 aforementioned molecules, λ+ and λ−, were calculated using (a) the energy of the optimized neutral and cationic structures, (b) the energy of the cation species with the geometry of the neutral species, and (c) the energy of the neutral species with the geometry of the cation species, as shown in Table 2. These calculations were performed at the B3LYP/6-31+G(d,p), M06-2X/6-31+G(d,p), and wB97XD/6-31+G(d,p) levels of theory.
Organization energies λ (meV/mol) of 11 coronene derivatives at the different density functional methods.

When the B3LYP method is used to calculate the study molecule or ion-related energy, the delocalization effect will be overestimated, resulting in the calculation that the resulting hole and electron transport recombination energies are too small. Based on the data shown in Table 2, the B3LYP method gave the lowest reorganization energies for hole and electron transport, whereas the long-range-corrected wB97XD method gave the highest energies. All three methods produced the same trends in terms of the relative hole and electron transport reorganization energies for each molecule. The hole transport reorganization energies (λ+) obtained for
Charge transport matrix elements and rate constants
When studying the carrier mobility of molecules, it is necessary to calculate the energies corresponding to different rotational angles between the neutral and ionic species of a dimer, Ei, and also the corresponding energy gap between HOMO and HOMO – 1, and LUMO and LUMO + 1. These calculations will yield the matrix elements t of hole and electron transport. In order to simplify the model, the dimers were not further structurally optimized. We carried out these calculations with angles ranging from 0° to 180°, in 20° intervals. The charge transport matrix elements characterize the coupling strength of electron–electron interactions, which is a major factor for carrier mobility and charge transport rates. The value of the matrix element increases as coupling strength increases, and hence so does charge mobility; high coupling strengths are therefore beneficial for charge transport. To facilitate this analysis, Figure 4 illustrates the frontier diagram corresponding to different rotational angles between the neutral and ionic species of a dimer that were obtained by stacking stable

The frontier molecular orbital of molecule
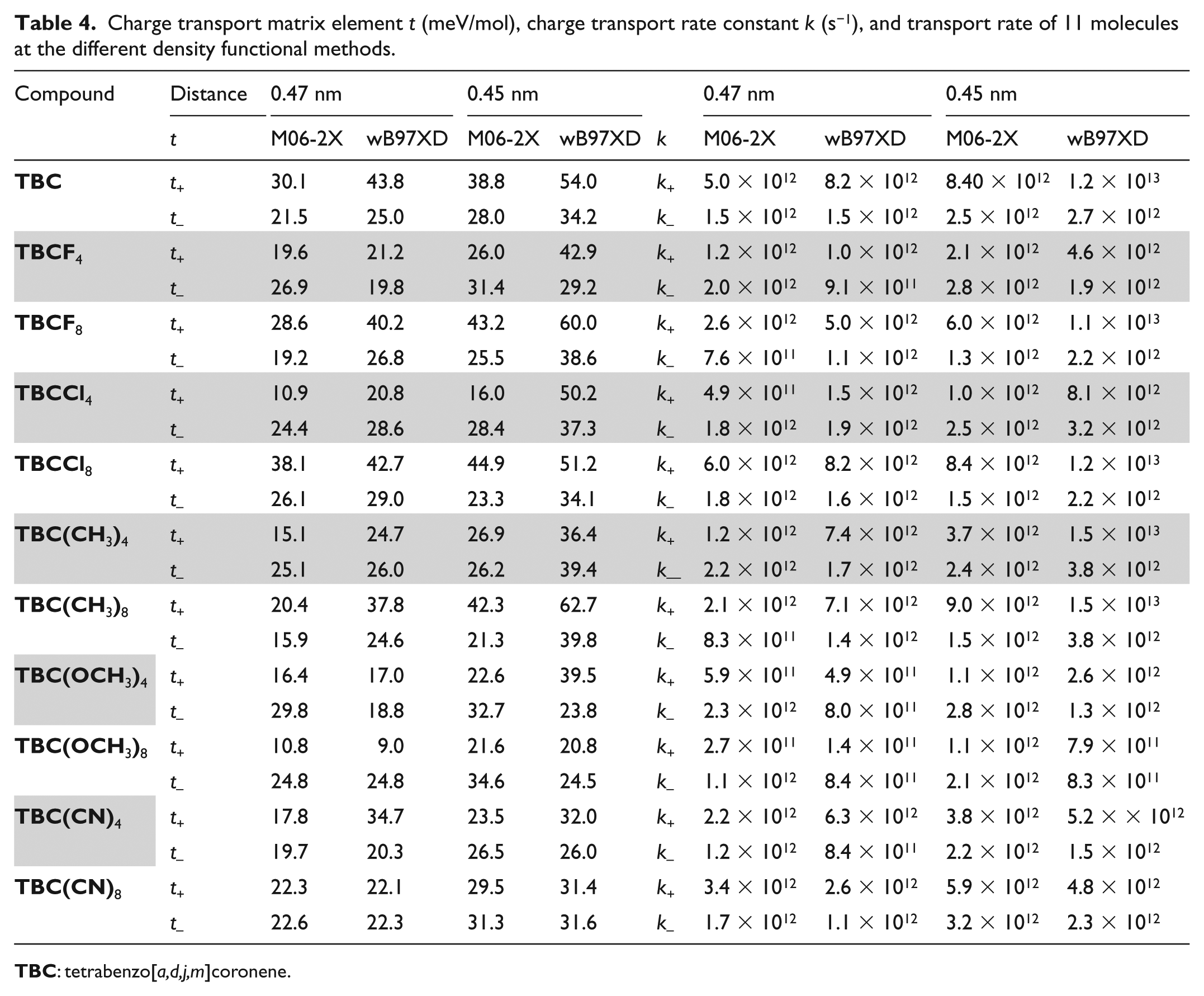
Since the 11 benzocoronene molecules included in this study are large quasi-planar conjugated π systems, an excessively small centroid distance between the monomers of a dimer would lead to overlaps between the side groups of the neutral and ionic species, whereas an excessively large centroid distance would significantly weaken their interactions and reduce carrier mobility values. Table 3 shows the charge transport matrix elements and rate constants of hole and electron transport in a dimer with centroid distances ranging from 0.45 to 0.50 nm, calculated at the B3LYP/6-31+G(d,p) level of theory. The data in Table 3 indicate that the charge transport matrix elements and rate constants of all 11 molecules generally increased with decreasing centroid distance. The differences between the results corresponding to centroid distances of 0.50 and 0.45 nm were significant, whereas most of the calculated results with centroid distances of 0.47 and 0.45 nm were relatively similar. To obtain a suitable method for calculating the carrier mobility of TBC-like molecules and thus reduce computational workloads, we used M06-2X and wB97XD functionals with dimer centroid distances of 0.47 and 0.45 nm. The hole and electron transport matrix elements and charge transport rates calculated for the 11 TBC variants are shown in Table 4. From the comparison of data in Tables 3 and 4, it can be seen that the wB97XD method uses the DFT-D2 dispersion correction and long-range correction to calculate the weak interaction between dimers. The hole and electron transport matrix obtained from the calculation of the center-to-center distance of the same dimer is significantly larger than that obtained by the B3LYP method, therefore impairing the effect of the increase in recombination energy on the decrease in the molecular carrier transport rate. The result of the synergistic effect makes the hole/electron transport matrix elements and rate constants calculated using wB97XD and B3LYP methods relatively similar, whereas the results of the calculations of the M06-2X method deviate toward lower values. For the 11 TBC variants studied in this work, all three methods resulted in identical qualitative trends.
Charge transport matrix element t (meV/mol) and charge transport rate constant k (s−1) of 11 molecules at the B3LYP/6-31+G(d,p) level.

Charge transport matrix element t (meV/mol), charge transport rate constant k (s−1), and transport rate of 11 molecules at the different density functional methods.
Carrier mobility
The B3LYP method overestimates the delocalization effect. Compared with the B3LYP method, the variant hybrid functional M06-2X method takes into account the weak intermolecular interactions between dimers. The long-range-corrected functional wB97XD method calculating the weak interaction between dimers is considered both the DFT-D2 dispersion correction and the long-range correction, which is more conducive to improving the accuracy of the weak interaction between dimeric molecules. From Table 2, we can see that the B3LYP method calculates the reorganization energy data obtained, which are significantly greater than the results obtained by the M06-2X or wB97XD method. From the comparison between Tables 3 and 4, we know that the B3LYP method calculates the resulting charge transfer matrix, which is much smaller than the calculated value by the M06-2X or wB97XD method. The result of the above synergistic effect makes the carrier mobility obtained by the calculation of M06-2X and wB97XD very similar.
The carrier mobilities calculated at the B3LYP/6-31+G(d,p), M06-2X/6-31+G(d,p), and wB97XD/6-31+G(d,p) levels of theory using different centroid distances are shown in Table 5. In this table, it is shown that the hole transport rates of
Transport rate μ (cm2 V−1 s−1) of 11 molecules using B3LYP/6-31+G(d,p), M06-2X/6-31+G(d,p), and B97XD/6-31+G(d,p) levels.

All three methods exhibited the same trends: for–F-, –Cl-, –CH3-, and –CN-substituted
Conclusion
In this work, we used DFT functionals such as B3LYP, M06-2X, and wB97XD with the 6-31+G(d,p) basis set to perform geometry optimizations and charge transport rate calculations on 11 variants of
Supplemental Material
SI2018 – Supplemental material for Theoretical investigations on charge transport properties of tetrabenzo[a,d,j,m]coronene derivatives using different density functional theory functionals (B3LYP, M06-2X, and wB97XD)
Supplemental material, SI2018 for Theoretical investigations on charge transport properties of tetrabenzo[a,d,j,m]coronene derivatives using different density functional theory functionals (B3LYP, M06-2X, and wB97XD) by Ziran Chen, Yuan Li, Zhanrong He, Youhui Xu and Wenhao Yu in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Technology Department of Sichuan Province (grant number 2015GZ0343) and the Project of Sichuan Provincial Department of Education (grant numbers 16ZB0059, 17ZA0346).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.