Treatment of acridine with 1-aminobenzotriazole and lead tetraacetate gives N-phenylacridone by a non-concerted benzyne reaction. A mechanism is proposed by which an initial adduct is converted into this product.
The oxidation of 1-aminobenzotriazole 1 with lead tetraacetate by the method of Campbell and Rees allows the rapid generation of benzyne 2 under mild conditions (Figure 1).1–3
The oxidation of 1-aminobenzotriazole 1 with lead tetraacetate.
In the absence of other intercepting reagents, biphenylene 3 is formed with a minor amount of triphenylene 4. Of interest to us were the failed attempts to react benzyne 2 with either acridine 5 or phenazine 7 to give azatriptycene 6 and diazatriptycene 8, respectively (Figure 2).2 Adducts 6 and 8 are interesting targets for synthesis2 because of their triptycene-derived shape and the basic nitrogens arising from their relationship to the three benzene rings.
Failed attempts to react benzyne 2 with acridine 5 and phenazine 7.2
Benzyne 2 has a high reactivity and so we thought that these reactions should work by a concerted or a non-concerted reaction mechanism. Campbell and Rees did report an unknown compound from the lead tetraacetate oxidation of 1-aminobenzotriazole 1 in the presence of acridine 5 but it was not characterised, only given a melting point (m.p.) of 270–271 °C.2 This is above the m.p. of acridine 5 (107–110 °C). The reaction with phenazine 7 was reported to give a complex mixture of red, blue and purple crystals.2 Azatriptycene 6 has been prepared by a tandem benzyne method.4 The oxidation of 1-aminobenzotriazole 1 with lead tetraacetate has been widely studied for the generation of benzyne intermediates.5–8
Results and discussion
The experimental method for the lead tetraacetate oxidation of 1-aminobenzotriazole in the presence of acridine is straightforward, but the chromatographic purification of products is not. Biphenylene 3 is easily separated from the reaction, but care must be taken to isolate a low yield of a further product and separate it from an excess of acridine 5. With care though this can be done to give a pure product which has two infrared (IR) stretches, 1633 and 1598 cm−1, rather than a typical carbonyl group stretch at 1700 cm−1. The spectroscopic data suggested structure 9 and this was confirmed by an X-ray single-crystal structure determination.9 Structure 9 has been reported previously but was prepared by a different method.9 Data on related compounds is also available.10–12 The two low IR stretches are presumably because of strong resonance conjugation from the amine lone pair to the carbonyl group. The m.p. of 268–269 °C is similar to the value of 270–271 °C reported by Campbell and Rees2 suggesting that these compounds are the same (Figures 3 and 4).
The reaction of benzyne 2 with acridine 5 to give N-phenylacridone 9.
Crystallographic drawing of N-phenylacridone 9.
The acridone ring system in 9 shows a small, but significant, pucker (dihedral angle between the C1 and C7 rings = 2.22 (5)°; dihedral angle between the C7 and C9 rings = 5.32 (5)°). The dihedral angle between the tricyclic ring system and the pendant phenyl group is 75.92 (4)°. In the crystal, inversion dimers are linked by pairs of C15–H15⋯O1 (H⋯O = 2.38 Å) hydrogen bonds.
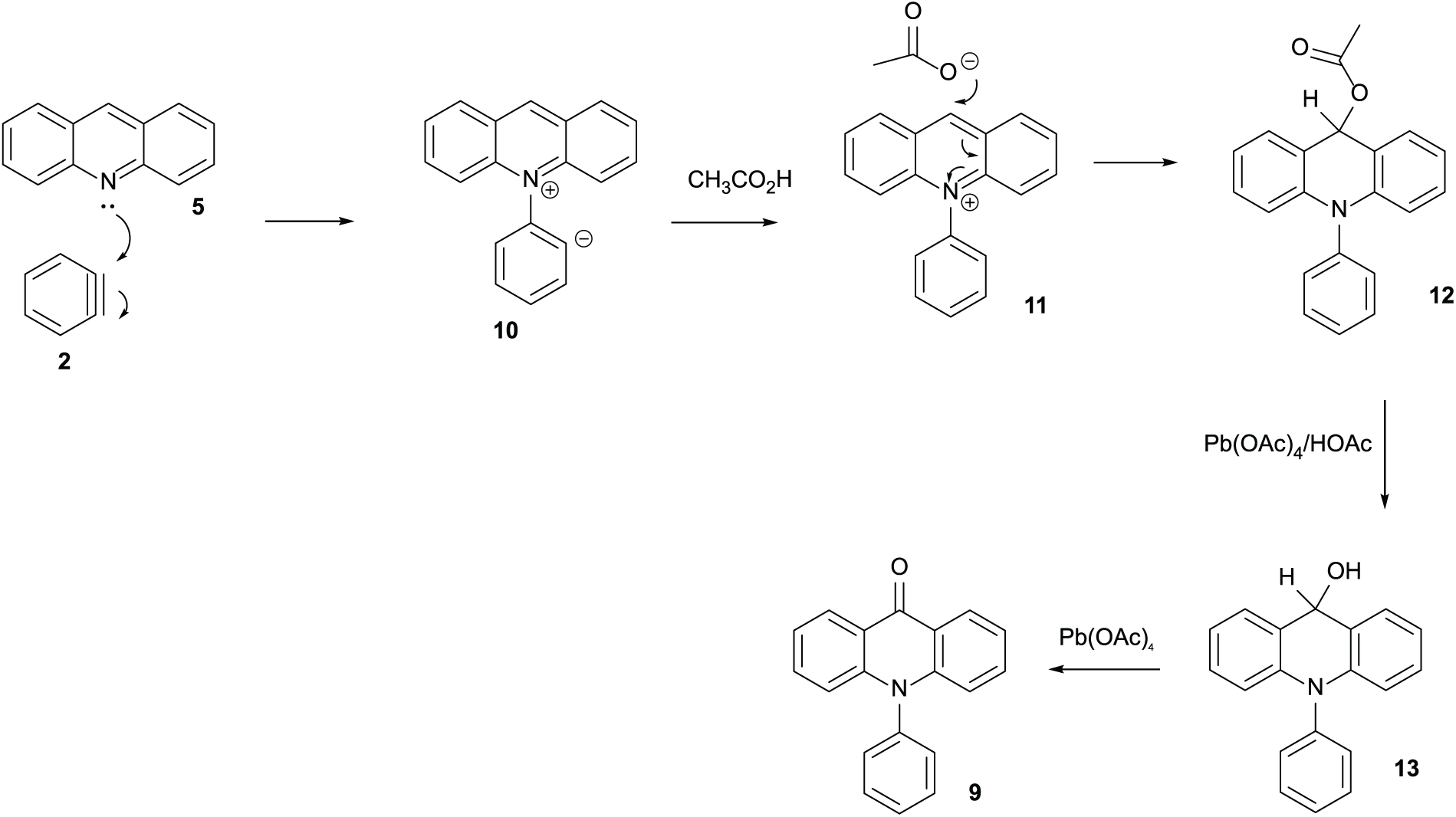
Figure 5 shows a proposal for how this unusual compound might have been formed. Benzyne 2 appears not to have undergone a concerted cycloaddition with acridine 5. Instead, a non-concerted reaction has occurred because the acridine nitrogen is nucleophilic leading to ylide 10 then to salt 11 because there is acetic acid present in lead tetraacetate. The counter-ion must be acetate and might add as a nucleophile to the N-phenylacridinium salt 11 to give acetate 12. If acetate 12 hydrolyses to alcohol 13, the secondary alcohol could be oxidised by more lead tetraacetate to the final product N-phenylacridone 9.
A possible route for the formation of N-phenylacridone 9.
No products were obtained from reacting benzyne 2 with phenazine 7, with or without the addition of NaBH4 to reduce an intermediate phenazinium salt, so this reaction remains elusive.
Summary
A study of the reaction of benzyne 2 with acridine 5 has revealed that the mystery product formed is N-phenylacridone 9. Benzyne 2 reacts by a non-concerted mechanism rather than by a concerted mechanism owing to the nucleophilic nitrogen of acridine 5 and its poor reactivity as an inverse electron-demand diene. The m.p. of 268–269 °C for compound 9 is close to that reported by Campbell and Rees (270–271 °C) suggesting that this is the same compound which has now been identified.
Experimental
IR spectra were recorded on an ATI Mattson Fourier transform infrared (FTIR) spectrometer using KBr discs. Ultraviolet (UV) spectra were recorded using a Perkin-Elmer Lambda 25 UV-Vis spectrometer with CH2Cl2 as the solvent. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 and 100.5 MHz, respectively, using a Varian 400 spectrometer. Chemical shifts, δ, are given in ppm and measured by comparison with the residual solvent. Coupling constants, J, are given in Hz. Low- and high-resolution mass spectra were obtained at the University of Wales, Swansea using electron impact ionisation and chemical ionisation. Melting points were determined on a Kofler hot-stage microscope.
Synthesis of N-phenylacridone 9
1-Aminobenzotriazole 1 (50 mg, 0.37 mmol) and acridine 5 (67 mg, 0.37 mmol) were stirred in dichloromethane (DCM; 50 mL) with Pb(OAc)4 (165 mg, 0.37 mmol) for 24 h. The mixture was poured directly onto a flash silica column (12 in × 1 in) and eluted with DCM to remove biphenylene 3. Elution with ether:DCM (10:90) and careful fraction collection from the excess acridine 5 gave the title compound (5.0 mg, 5.0%) as a yellow solid, m.p. 268–269 °C (from DCM:light (40–60 °C) petroleum ether). λmax (EtOH)/nm 395 (log ε 3.5), 380 (3.4) and 250 (4.1); νmax (KBr)/cm 3047w, 1633s, 1598s, 1489s, 1458s, 1359s, 1305s, 1162m, 1038w, 942w, 934w, 778m, 748s, 705s, 672s, 620s, 558s and 514; δH (400 MHz; CDCl3) 6.69 (2H, d, J = 8.0), 7.21 (2H, t, J = 6.0 and 8.0), 7.30 (2H, d, J = 8.0), 7.43 (2H, t, J = 6.0 and 8.0), 7.64 (1H, t, J = 4.0 and 8.0), 7.64 (2H, t, J = 8.0 and 8.0) and 8.52 (2H, d, J = 8.0); δC (100.1 MHz; CDCl3) 116.9, 121.6, 121.8, 127.3, 129.6, 130.1, 131.1, 133.3, 138.9, 143.2 and 178.2; m/z (Orbitrap ASAP) 272.1062 (M+ + H, 100%) C19H14NO requires 272.1070.
Crystal structure
Colourless plates of 9 were recrystallised from DCM/light petroleum ether solution. Intensity data were collected at T = 100 K using a Rigaku Mercury CCD diffractometer (Mo Kα radiation, λ = 0.71073 Å): C19H13NO, Mr = 271.30, triclinic, space group (No. 2), a = 8.5687 (5), b = 9.0551 (4), c = 9.3238 (4) Å, α = 68.028 (4), β = 78.202 (5), γ = 76.553 (5)°, V = 646.94 (6) Å3, Z = 2, R(F) = 0.036, wR(F2) = 0.106, CCDC deposition no. 1883546.
Footnotes
Acknowledgements
We are grateful to the UK EPSRC National Mass Spectrometry Service Centre for mass spectrometric data.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental Material
CCDC1883546 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via .