Fmoc Rink linker is a widely used peptide-resin linker in the solid-phase synthesis of peptide-amides. This paper describes an improved and practical eight-step synthetic approach for this compound in a 50% overall yield, using p-hydroxybenzaldehyde as the starting material. The procedure is operationally simple and amenable to scale-up synthesis.
Since Merrifield first developed solid-phase peptide synthesis,1,2 this technology has caused a paradigm shift within the peptide synthesis community. Peptide chains can be rapidly assembled through successive reactions of amino acids onto an insoluble porous support. So far, solid-phase synthesis has been widely applied for the preparation of a variety of peptides.
In solid-phase peptide synthesis, the solid support commonly consists of polymeric resin beads, which are functionalised with reactive linkers. Most linkers are bifunctional spacers, with one end permanently anchoring to the resin and the other end temporarily linking to the peptide (Figure 1). Rink linker was first designed by Rink and further developed by Bernatowicz and co-workers for the preparation of peptide C-terminal amides.3,4 Peptides anchoring to Rink linker can be easily cleaved under acidic conditions (Figure 1). Since the discovery of Rink linker, it has been grafted to a variety of resins and widely used for scientific investigations and industrial solid-phase peptide synthesis.5–7 Thus, Fmoc Rink linker (1) is the key reagent to prepare this series of Rink-resins.
Solid-phase strategy and Fmoc Rink linker (1).
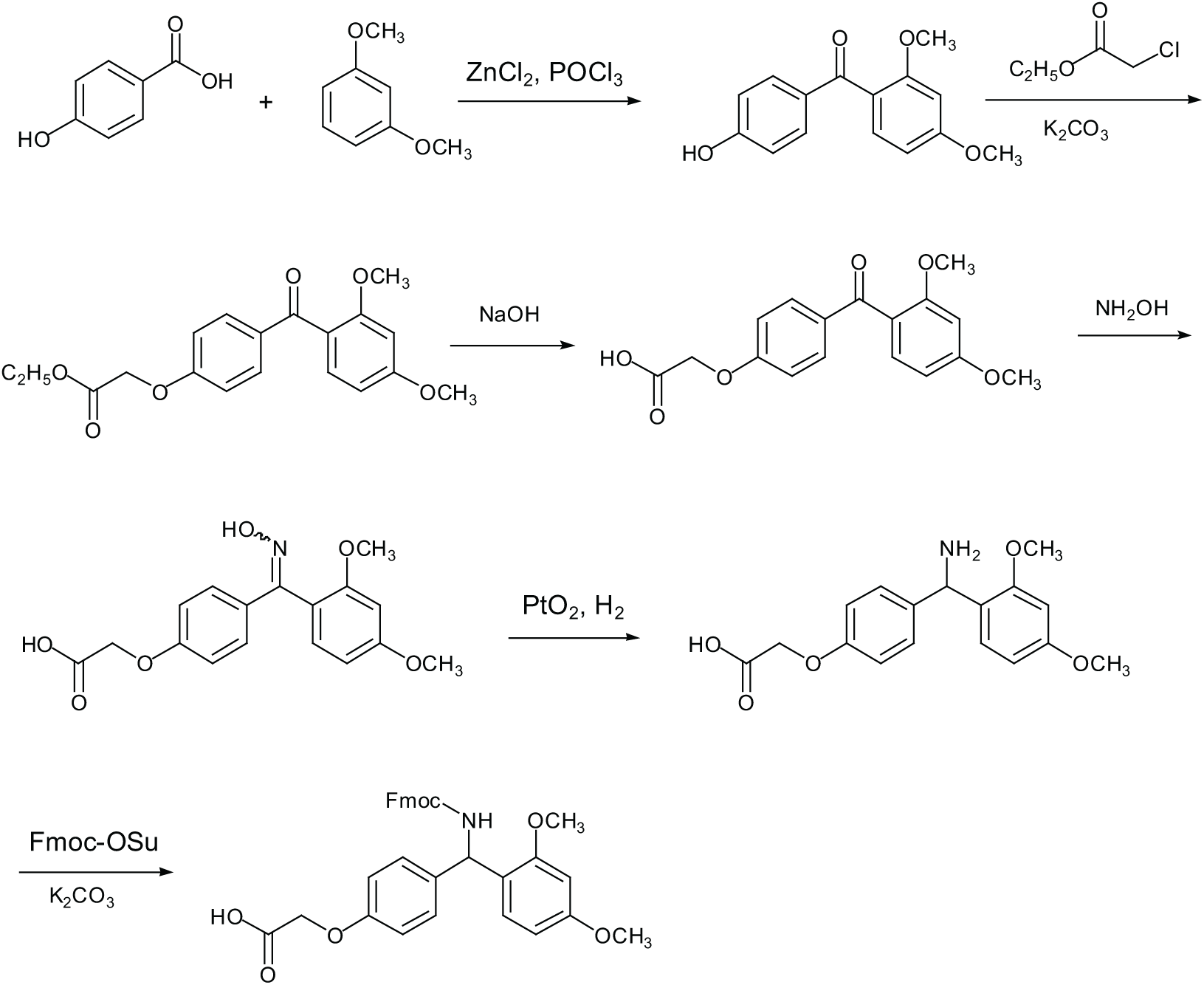
As far as we know, the synthetic routes for this linker are very few. Takahashi and co-workers reported a detailed synthesis of Fmoc Rink linker (1) in six steps, using 4-hydroxybenzoic acid and 1,3-dimethoxybenzene as the starting materials (Scheme 1).8 Although this synthetic route is relatively short and efficient, three major drawbacks exist in this protocol: (1) the Friedel–Crafts acylation of 1,3-dimethoxybenzene with 4-hydroxybenzoic acid produces a very impure product, (2) the use of platinum oxide to reduce the oxime is neither economical nor amenable to large scale synthesis and (3) the reduction of the oxime is time-consuming (two days!). Actually, we have repeated this Friedel–Crafts acylation of 1,3-dimethoxybenzene with 4-hydroxybenzoic acid. The purity of the product was poor. In order to obtain pure product, repeated recrystallisations were needed, resulting in a low yield (about 35%). The poor purity could be ascribed to the unprotected hydroxyl group. Thus, a more practical and economical synthetic strategy is needed to produce this peptide-resin linker.
In recent years, we have developed an improved preparation of Fmoc Rink linker (1) in seven steps, using p-hydroxybenzaldehyde as the starting material. This procedure has been patented by us and written in Chinese.9,10 Here we report our investigations in more detail.
Results and discussion
Our synthesis of Fmoc Rink linker (1) is outlined in Scheme 2. The starting material p-hydroxybenzaldehyde was treated with ethyl bromoacetate under basic conditions to give the aldehyde 2 in excellent yield. Subsequently, oxidation of the aldehyde 2 proceeded smoothly to afford the acid 3 in almost quantitative yield. Thereafter, the acid 3 was treated with SOCl2 to give the acyl chloride 4, which was directly used in the next step without purification.
The new synthetic pathway.
With the acyl chloride 4 available, we screened for suitable Friedel–Crafts acylation conditions. As shown in Table 1, among the four Lewis acids tested, AlCl3 was most effective in accomplishing the reaction. Among the four solvents tested, dichloromethane was the best, while trichloroethylene resulted in no products (Table 1, entry 8). The amount of the Lewis acid was also screened. It was found that stoichiometric Lewis acid (1.1 equiv.) was needed to ensure a good yield (Table 1, entry 4). However, more Lewis acid was not necessary (Table 1, entry 10).
Subsequently, the compound 5 was hydrolysed to give the acid 6. The direct reductive amination of 6 was attempted, but failed to give the desired product 8. Thus, we had to condense 6 with hydroxylamine and try to optimise the reduction conditions.
Although a number of articles have reported the reduction of oximes to amines,11–16 the reported methods for the specific compound 7 are few. To the best of our knowledge, only Takahashi and co-workers reported the reduction of this oxime 7 by hydrogenation.8 The use of platinum oxide and the long hydrogenation time (two days) made this protocol not practical for scale-up preparations. Besides, the carboxylic acid group may also interfere with the reduction reaction. Thus, optimised reduction conditions were needed for the reduction of the oxime, while retaining the carboxylic acid functionality. In addition, the desired product 8 posed several problems, owing to its polyfunctional and amphiprotic nature and poor solubility.
To screen for an operationally simple and cost-effective protocol, several conditions were investigated (Table 2). The hydrogenations catalysed by Pd/C or Raney Ni were first investigated but, unfortunately, failed to give the desired product (Table 2, entries 1 and 2). Since Zn/AcOH reduction has also been disclosed in several reports,17–19 we attempted this protocol and obtained the product in moderate yields (Table 2, entries 3 and 4). Further investigations indicated that Zn/NH3 reduction was much better than Zn/AcOH, producing 8 in a good yield (Table 2, entry 5).20 At last, the target compound 1 was obtained by the condensation of the amine 8 with N-(9-fluoroenylmethoxy-carbonyloxy)succinimide (Fmoc-OSu) in a good yield.
Hydrogenation at room temperature and 1 bar pressure.
No products obtained.
In conclusion, we describe an improved and practical eight-step synthetic approach for Fmoc Rink linker (1) in 50% overall yield, using p-hydroxybenzaldehyde as the starting material. The advantages of this procedure include simple operations, low production costs and good yields.
Experimental
Commercial reagents were used without further purification. The boiling range of petroleum ether used was 60–90ºC. Melting points were measured on a SGW X-4 (INESA) temperature apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker DRX-400 (400 MHz) spectrometer or a Bruker DRX-500 (500 MHz) spectrometer. 13C NMR spectra were obtained on a JNM-EX400 (100 MHz) spectrometer. Mass spectra (MS) were determined on a Bruker MicroTof II mass spectrometer or a Waters High Resolution UPLC-TOFMS spectrometer. Elemental analyses were performed using a VARIO EL III analyser. IR spectra were obtained using KBr disks on a Bruker Tensor 27 FTIR spectrometer.
Synthesis of (4-formylphenoxy)acetic acid ethyl ester (2)
Potassium carbonate (15 g, 0.11 mol) and ethyl bromoacetate (15.0 g, 89.8 mmol) were added to a solution of 4-hydroxybenzaldehyde (10.0 g, 82.0 mmol) in acetone (50 mL). The mixture was stirred at 60ºC for 3 h. Then the resulting mixture was filtered and the filtrate was concentrated and extracted with dichloromethane (150 mL). The organic phase was washed with saturated Na2CO3 solution (100 mL) and water (100 mL) and evaporated to give the crude product, which was triturated with petroleum ether to give the pure compound 2 as a colourless solid: Yield 16.7 g (98%); m.p. 35–37ºC (lit.21 32–34ºC); 1H NMR (CDCl3, 500 MHz): δ 9.83 (s, 1H), 7.73–7.83 (m, 2H), 6.90–6.98 (m, 2H), 4.63 (s, 2H), 4.21 (q, J = 7.1 Hz, 2H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 189.7, 167.0, 161.6, 130.9, 129.7, 113.9, 76.4, 76.1, 75.8, 64.2, 60.6, 13.1; IR (KBr): 2987, 2750, 1742, 1675, 1600, 1207, 1080, 840 cm–1.
Synthesis of 4-ethoxycarbonylmethoxybenzoic acid (3)
The aldehyde 2 (15.0 g, 72.1 mol) was dissolved in acetonitrile (30 mL). Then water (25 mL), potassium dihydrogen phosphate (19.0 g, 0.140 mol) and 30% hydrogen peroxide (12.0 g, 0.106 mol) were added to the solution. A solution of 80% NaClO2 (12.0 g, 0.106 mol) in water (25 mL) was added dropwise at 5–10ºC to the mixture which was then stirred at room temperature for 1 h. Thereafter, aqueous Na2SO3 was added slowly to the mixture to quench the reaction, until potassium iodide starch paper did not turn purple. The resulting mixture was acidified with concentrated hydrochloric acid to pH = 1. The precipitate was collected and dried to give the crude product, which was triturated with a mixture of petroleum ether and ethyl acetate (petroleum ether/ethyl acetate = 5:1) to afford the pure compound 3 as a colourless solid: Yield 15.3 g (95%); m.p. 141–142ºC (lit.22 138ºC); 1H NMR (DMSO-d6, 500 MHz): δ 7.85–7.91 (m, 2H), 6.98–7.03 (m, 2H), 4.87 (s, 2H), 4.18 (q, J = 7.1 Hz, 2H), 1.21 (t, J = 7.1 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz), δ 168.3, 166.9, 161.1, 131.2, 123.7, 114.3, 64.6, 60.7, 14.0; HRMS (ESI) m/z calcd for C11H12NaO5: [M + Na]+: 247.0577; found: 247.0598; IR (KBr): 3473, 2987, 1748, 1460, 1100, 840 cm–1.
Synthesis of [4-(2,4-dimethoxybenzoyl)phenoxy]acetic acid ethyl ester (5)
DMF (0.24 g, 3.3 mmol) and thionyl chloride (12.0 g, 0.101 mmol) were added to a suspension of the compound 3 (15.0 g, 67.0 mmol) in dichloromethane (100 mL). The mixture was refluxed for 2 h, and evaporated to dryness to give the acyl chloride 4, which was used immediately in the next step without purification. The acyl chloride 4 and 1,3-dimethoxybenzene (9.20 g, 66.7 mmol) were dissolved in dry dichloromethane (100 mL). Aluminum chloride (9.80 g, 73.7 mmol) was added slowly to the mixture at 0–5ºC. The reaction was stirred at 5ºC for 1 h. Then cold water (80 mL) was added dropwise to the mixture to quench the reaction. The organic layer was washed with aqueous NaHCO3 (80 mL) and water (80 mL), dried over Na2SO4, and evaporated under reduced pressure to give the crude product. This was recrystallised from a mixture of ethanol and H2O (ethanol/water = 1:1) to give a colourless solid 5: Yield 19.8 g (86%); m.p. 81–82ºC (lit.8 82–83ºC); 1H NMR (CDCl3, 500 MHz): δ 7.68–7.73 (m, 2H), 7.27 (d, J = 8.4 Hz, 1H), 6.79–6.86 (m, 2H), 6.42–6.49 (m, 2H), 4.60 (s, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.64 (s, 3H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 193.2, 167.3, 162.0, 160.2, 158.2, 131.4, 131.1, 130.7, 120.8, 112.9, 103.5, 97.8, 64.2, 60.5, 54.6, 54.5, 13.1; HRMS (ESI) m/z calcd for C19H20NaO6: [M + Na]+: 367.1152; found: 367.1168; IR (KBr): 3001, 1763, 1678, 1600, 1256, 1037, 848 cm–1.
A mixture of compound 5 (15.0 g, 43.6 mmol), water (15 mL) and sodium hydroxide (2.60 g, 65.0 mmol) in ethanol (40 mL) was stirred at room temperature for 2 h. Then ethanol was removed under reduced pressure. To the residue was added water (60 mL) and concentrated hydrochloric acid (6 mL). The precipitate was collected and oven-dried to afford the acid 6 as a colourless solid: Yield 13.5 g (98%); m.p. 160–161ºC (lit.8 158–159ºC); 1H NMR (DMSO-d6, 500 MHz): δ 7.61–7.65 (m, 2H), 7.24 (d, J = 8.4 Hz, 1H), 6.96–7.01 (m, 2H), 6.61–6.70 (m, 2H), 4.76 (s, 2H), 3.84 (s, 3H), 3.67 (s, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 193.3, 169.8, 162.4, 161.6, 158.4, 131.4, 130.9, 130.5, 121.3, 114.1, 105.3, 98.7, 64.8, 55.6, 55.5; HRMS (ESI) m/z calcd for C17H16NaO6: [M + Na]+: 339.0839; found: 339.0872; IR (KBr): 3001, 2500, 1748, 1650, 1601, 1417, 939, 819 cm–1.
Synthesis of {4-[(2,4-dimethoxyphenyl)hydroxyiminomethyl]phenoxy}acetic acid (7): A mixture of the compound 6 (12.0 g, 38.0 mol), hydroxylamine hydrochloride (7.60 g, 0.110 mol) and triethylamine (21.0 mL, 0.151 mol) in ethanol (30 mL) was refluxed for 12 h. Then the reaction mixture was evaporated to dryness to give a residue. To the residue was added water (60 mL) and concentrated hydrochloric acid to adjust the pH to 1–2. The precipitate was collected and oven-dried. The crude product was further triturated with ethyl acetate to give the pure 7 as a colourless solid: Yield 11.0 g (88%); m.p. 161–162ºC; 1H NMR (DMSO-d6, 500 MHz, the main isomer): δ 13.00 (br, 1H), 11.09 (br, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 8.4 Hz, 2H), 6.52–6.61 (m, 2H), 4.67 (s, 2H), 3.79 (s, 3H), 3.55 (s, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 170.0, 160.9, 158.5, 157.4, 152.2, 131.3, 130.6, 127.2, 114.1, 113.4, 104.7, 98.9, 64.3, 55.4, 55.2; HRMS (ESI) m/z calcd for C17H18NO6: [M + H]+: 332.1129; found: 332.1131; IR (KBr): 2994, 2939, 1742, 1602, 1408, 1163, 935, 834 cm–1.
Synthesis of {4-[amino(2,4-dimethoxyphenyl)methyl]phenoxy}acetic acid (8): A mixture of compound 7 (8.0 g, 0.024 mol) and Zn powder (10.9 g, 0.168 mol) in concentrated ammonium hydroxide (60 mL) was stirred at 80ºC for 18 h. Then the insoluble solid was filtered off. The pH of the filtrate was adjusted to 7 by cautious addition of concentrated hydrochloric acid at room temperature. The resulting precipitate was collected and triturated with a mixture of ethanol and water (ethanol/water = 1:1) to give the product 8 as a colourless solid: Yield 6.5 g (86%); m.p. 216–217ºC (lit.8 215–217ºC); 1H NMR (500 MHz, D2O-NaOD): δ 6.81–6.87 (m, 3H), 6.51 (d, J = 8.7 Hz, 2H), 6.17–6.20 (m, 2H), 4.83 (s, 1H), 4.07 (s, 2H), 3.42 (s, 3H), 3.32 (s, 3H). Anal. calcd for C17H19NO5: C, 64.34; H, 6.03; N, 4.41; found: C, 64.17; H, 6.20; N, 4.24%; IR (KBr): 3163, 2931, 1650, 1404, 1045, 925, 840 cm–1.
Synthesis of Fmoc Rink linker (1)
A mixture of compound 8 (4.5 g, 14 mmol) and potassium carbonate (0.98 g, 7.0 mmol) in dioxane–water (30 mL, 2:1) was stirred at room temperature for 30 min. Then Fmoc-OSu (4.78 g, 14 mmol) was added. After stirring for 6 h at room temperature, the solution was adjusted to pH 1–2 with 1 M hydrochloric acid. Then the mixture was extracted with ethyl acetate (50 mL). The organic layer was washed with saturated brine (30 mL × 2), dried and concentrated to give the crude product, which was crystallised from ethanol to give a colourless solid 1: Yield 6.5 g (85%); m.p. 177–178.5ºC (lit.8 178–179ºC); 1H NMR (DMSO-d6, 500 MHz): δ 12.95 (br, 1H), 8.11 (d, J = 9.2 Hz, 1H), 7.88 (d, J = 7.5 Hz, 2H), 7.72 (d, J = 7.3 Hz, 2H), 7.39–7.43 (m, 2H), 7.24–7.34 (m, 3H), 7.09 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 6.51–6.56 (m, 2H), 6.07 (d, J = 9.2 Hz, 1H), 4.63 (s, 2H), 4.19–4.33 (m, 3H), 3.75 (s, 3H), 3.73 (s, 3H), 3.32 (br, 1H); HRMS (ESI) m/z calcd for C32H29NNaO7: [M + Na]+: 562.1836; found: 562.1867; IR (KBr): 3300, 2959, 1742, 1650, 1608, 1412, 1100, 920, 834 cm–1.
Supplemental Material
JCR1805766_ESI – Supplemental material for An improved and practical synthesis of Fmoc Rink linker
Supplemental material, JCR1805766_ESI for An improved and practical synthesis of Fmoc Rink linker by Ling-yan Qin, Hao-ting Zhu, Li Zhan, Yong Zhu and Yu Luo in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: We thank the Laboratory of Organic Functional Molecules, the Sino-French Institute of ECNU for support.
Supplemental material
Supplemental material for this article is available online.
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.