The bis-oxime of acenaphthenequinone and the mono-oxime of benzil have been sulfonated by reaction with 4-methylbenzenesulfonyl chloride and propylsulfonyl chloride. The four sulfonated oximes were characterised by X-ray single-crystal structure determinations. Some photochemical decompositions were studied using a 6-W 254-nm immersion well lamp in dichloromethane. The 4-methylbenzenesulfonate bis-oxime of acenaphthenequinone and the 4-methylbenzenesulfonate mono-oxime of benzil both give 4-methylbenzenesulfonic acid upon irradiation but not 4-methylbenzenesulfinic acid. Fragmentation pathways are discussed. The possible use of these compounds as photoacid generators in polymer resists and the role of secondary reactions to liberate acid is discussed.
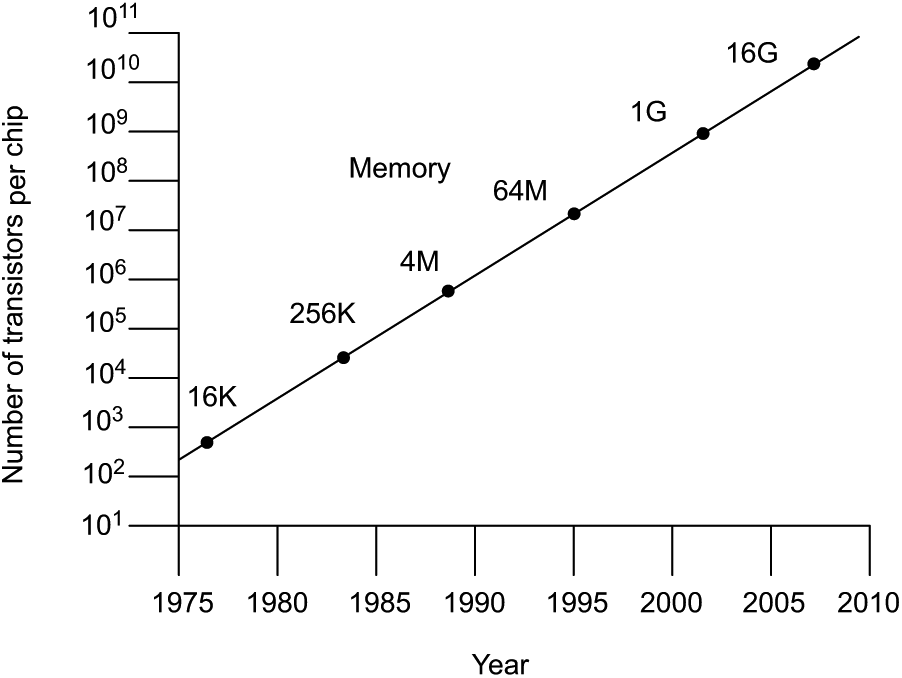
Improvements in the performance of semiconductor devices arise because of the decreasing size of the features on a silicon chip.1 Gordon E Moore,2 a co-founder of Intel, made the observation in 1965 that circuit densities of semiconductors would continue to double on a regular basis. This has become known as Moore’s Law and illustrates the astounding developments made in the field (Figure 1).1
A chart illustrating Moore’s Law.
Semiconductor devices or computer chips are fabricated by microlithography (Figure 2).1 In this technology, a radiation-sensitive polymer is spin coated and dried, forming a thin film of 1–0.1 μm thickness, on a single-crystal silicon wafer forming a resist. This is irradiated through a mask forming a pattern and then the exposed resist films are developed to create images. If the irradiated image is more soluble, it is classed as a positive system, and if it is less soluble, it is classed as a negative system. The remaining resist film serves as a protective layer during etching of the substrate. After etching, the remaining resist film is removed leaving behind a circuit pattern. The process is repeated to fabricate complex semiconductor devices.
The lithographic imaging process.
The resists contain a light-sensitive compound which upon irradiation and development modifies the solubility properties of the resist polymer (Figure 3).3,4 The success of the semiconductor industry’s recent developments has been due to the use of photoacid generators (PAGs) which liberate a small quantity of acid that catalyses a chemical reaction in a development step. For example, acid-catalysed deprotection of tert-butyl esters, liberating isobutene, leaves polymer-bound carboxylic acids which solubilise the polymer in aqueous base. Compounds 1 and 2 are likely to liberate the acid of a stable counter-anion,5–7 whereas compounds 3–5 will liberate a sulfonic acid.3,4,8–11 Decreasing feature size is commensurate with the use of higher energy radiation ranging from the ultraviolet (UV; 450–190 nm) down to extreme ultraviolet (EUV) at 7 nm.3,12–14
Some representative photoacid generators where R = different alkyl and aryl groups.
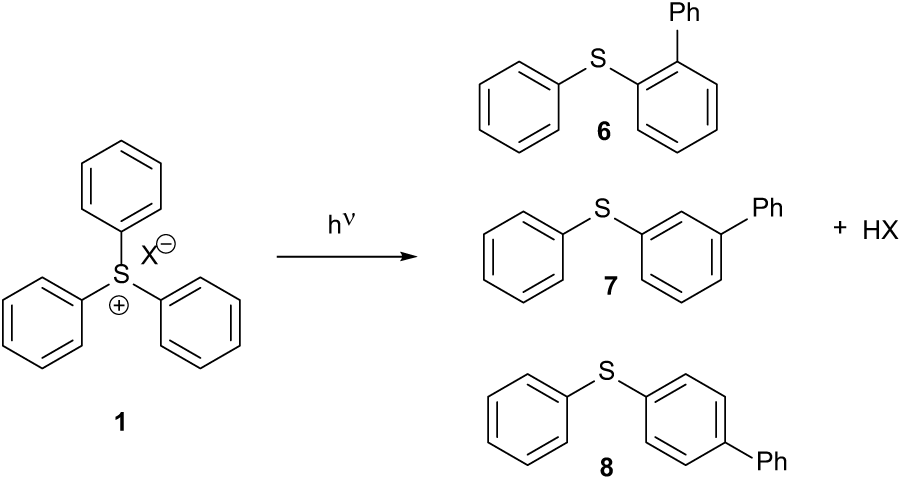
Figure 4 shows a possible mechanism for the photochemical fragmentation of a Crivello or triarylsulfonium salt.3 The non-nucleophilic counter-ion becomes the anion of a strong acid HX. A ring proton of the Crivello salt 1 is substituted for the phenyl ring and becomes the proton of the strong acid.
The photochemical fragmentation of a triphenylsulfonium salt to give a phenylthio-substituted biaryl and a strong acid.
The aim of this project is to develop an understanding of how the class of photoacid generators based on sulfonated oximes can function to modify polymer resists. Some compounds that are representative of examples in the literature4,8,10,11 have been prepared and their photochemical decomposition products studied.
Results and discussion
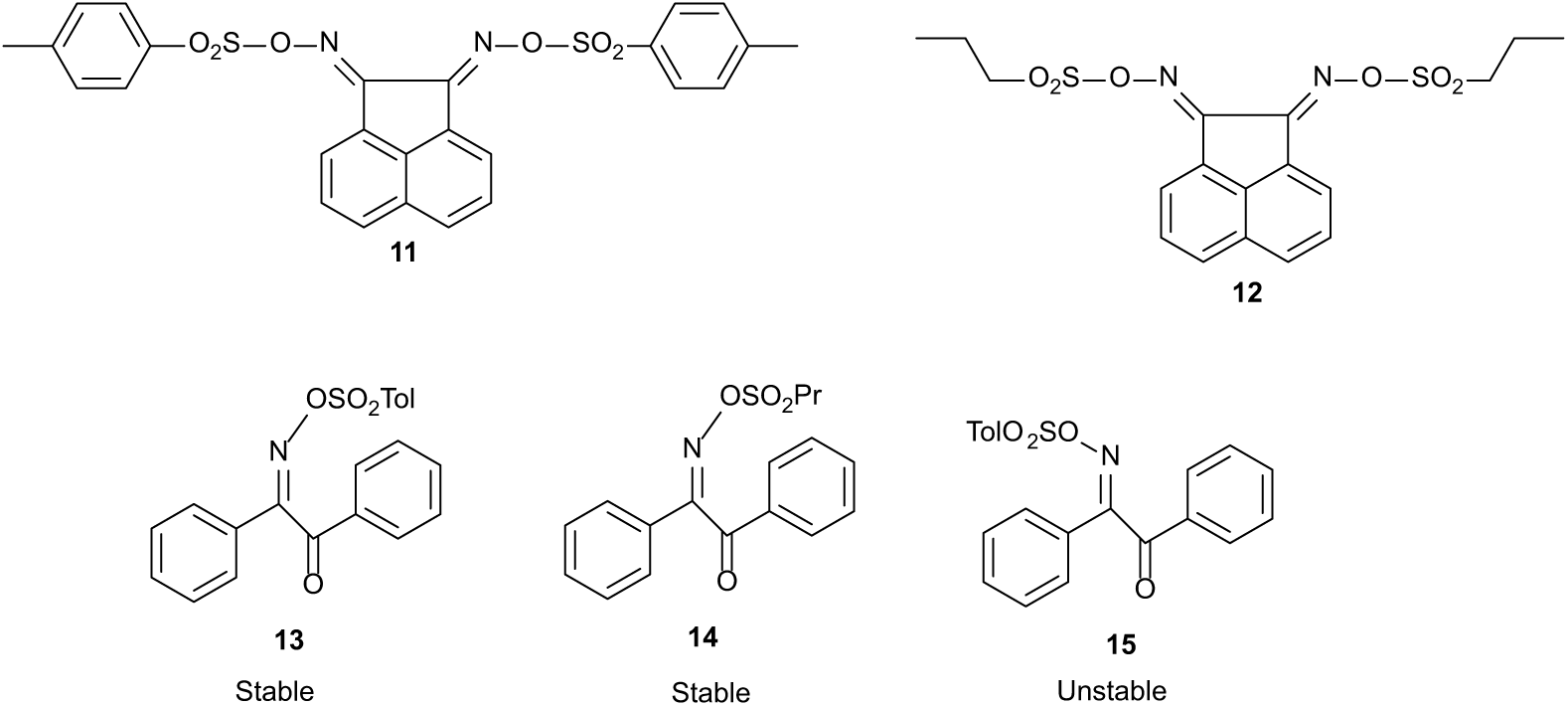
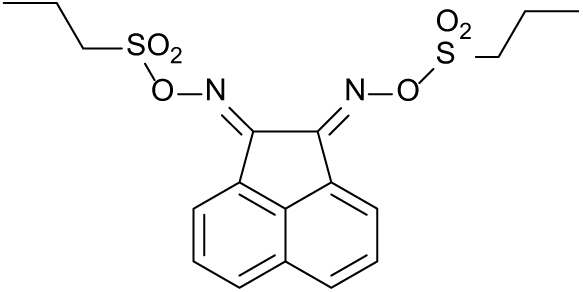
The condensation of NH2OH with acenaphthenequinone gives the known bis-oxime 915 and with benzil gives the known mono-oxime 1016 only and not a bis-oxime of benzil which is sometimes reported (Figure 5). There are a number of erroneous literature reports claiming that the bis-oxime of benzil can be formed under these conditions.17–19 Both syn and anti isomers of benzil derivative 10 have been claimed as they can be separated and the anti isomers form metal-ion complexes.16 We found that compounds 9 and 10 were both sulfonated with either 4-methylbenzenesulfonyl chloride or propylsulfonyl chloride to give compounds 11–14 which are potential photoacid generators (Figure 6). They have been characterised by X-ray single-crystal structure determination. The crystal structures show the stereochemistry of these compounds and that of the oximes from which they were made. Only one isomer was formed for each compound 11–14. Compounds 11 and 12 have both the sulfonate groups pointing away from each other which will arise for steric reasons. However, compounds 13 and 14 are syn isomers and are stable. According to the literature, the anti isomer 15 is unstable during synthesis for stereoelectronic reasons.16 It is made from the photochemically isolated anti oxime.16 The molecule fragments with the N-OSO2R group trans to the C–CO bond. In contrast to this, the stability of the syn isomers 13 and 14 is striking. The mono-oxime of benzil 10 initially forms as an oil but slowly crystallises to a white solid after a few hours and is a single isomer by 1H and 13C nuclear magnetic resonance (NMR).
Oximes of acenaphthenequinone 9 and benzil 10.
Oxime sulfonates 11–14 characterised by single-crystal X-ray structure determinations and a proposed unstable sulfonate 15.16 (Tol = 4-MeC6H4-, Pr = propyl-).
X-ray single-crystal structures
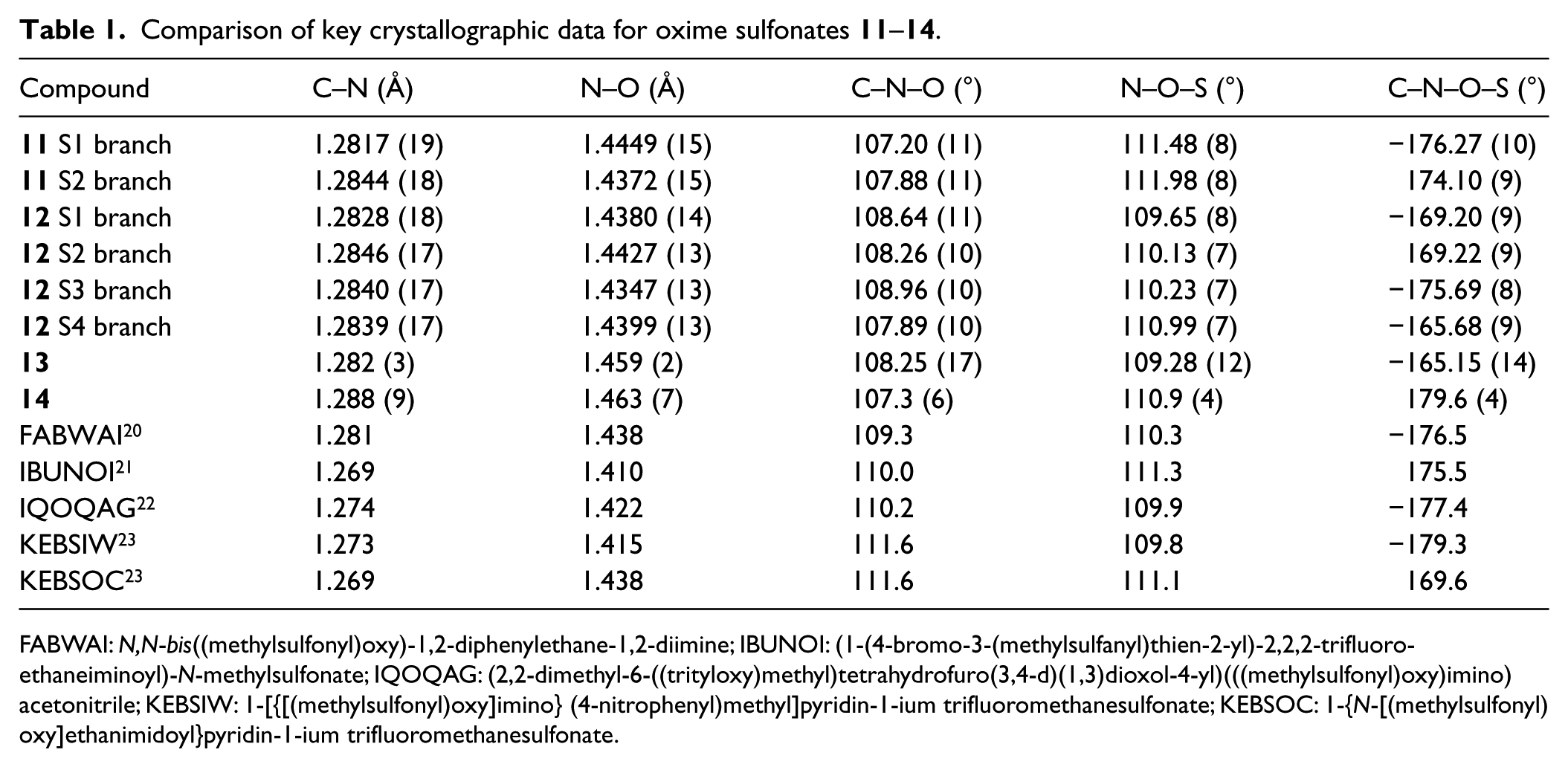
Key geometrical data for 11–14 and known oxime sulfonate crystal structures are compiled in Table 1.
Comparison of key crystallographic data for oxime sulfonates 11–14.
It may be seen that the C=N and N–O distances of the oxime groups in 11–14 and in other known oxime sulfonate crystal structures20–23 are all very consistent, as are the C=N–O and N–O–S bond angles. The C=N–O–S torsion angles indicate a preference for near planarity for these atoms, which is assumed to be the most stable conformation for oximes24 and any small deviations might be ascribed to packing forces in the crystal.
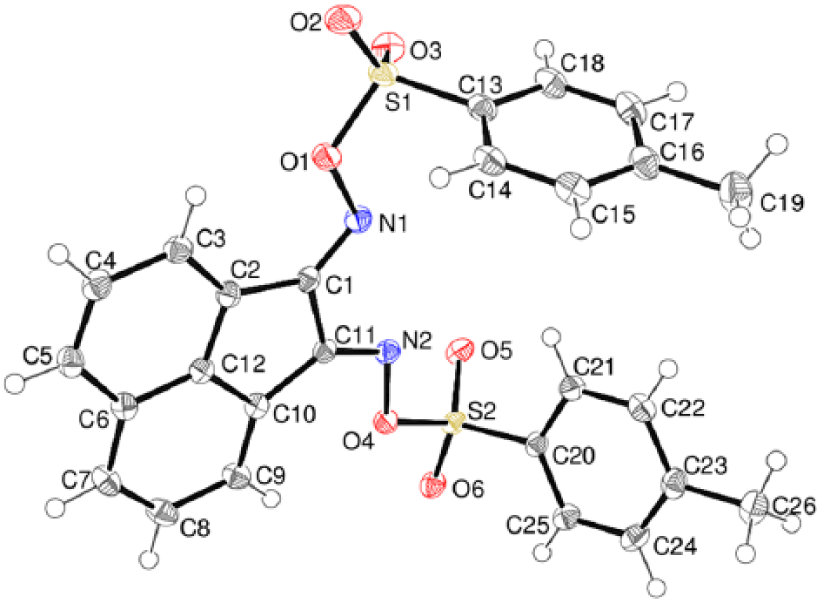
In compound 11 (Figure 7), the dihedral angles between the C1–C12 ring system and the pendant C13–C18 and C20–C25 phenyl groups are 81.49 (6)° and 66.93 (6)°, respectively. In the crystal of compound 11, the molecules are linked by weak interactions.
The molecular structure of compound 11 showing 50% displacement ellipsoids.
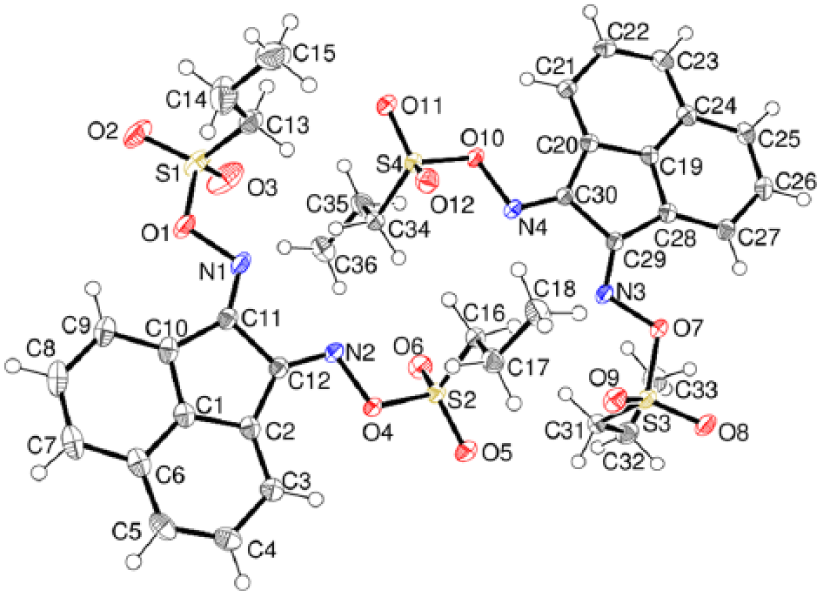
Compound 12 crystallises with two molecules in the asymmetric unit (Figure 8) with very similar geometries apart from the propyl chains of the sulfonate groups. In the S1 molecule, both of these adopt anti conformations [S1–C13–C14–C15 = −178.27 (13)°; C2–C16–C17–C18 = 171.17 (10)°], whereas in the S3 molecule one is gauche and one is anti [S3–C31–C32–C33 = −60.66 (15)°; S4–C34–C35–C36 = −171.53 (11)°]. In the crystal of compound 12, the molecules are linked by weak and interactions.
The molecular structure of compound 12 showing 50% displacement ellipsoids.
In compound 13 (Figure 9), the dihedral angles involving the C10–C16 ring (A), the C16–C21 ring (B) and the C1–C6 ring (C) are A/B = 87.28 (11)°, A/C = 50.74 (11)° and B/C = 44.88 (11)°. The N1–C8–C9=O4 torsion angle is −94.6 (2)°. In the crystal of compound 13, the molecules are linked by weak interactions.
The molecular structure of compound 13 showing 50% displacement ellipsoids.
In compound 14 (Figure 10), the dihedral angle between the C1–C6 and C9–C14 benzene rings is 77.3 (2)° and the propyl chain adopts an extended conformation [S1–C15–C16–C17 = 176.1 (6)°]. In the crystal of compound 14, the molecules are linked by weak interactions.
The molecular structure of compound 14 showing 30% displacement ellipsoids.
Photochemical irradiation
Compounds 11 and 13, representative of many other compounds,4,8,10,11 were irradiated in CH2Cl2 with a 6-W 254-nm lamp in a 100-mL immersion well for 5 h. This was done without deoxygenation because some polymer resist films are irradiated in air (365 nm i line, 248 nm KrF laser and 193 nm ArF laser by a dry process). Thin-layer chromatography (TLC) analysis of the mixture after evaporation of the solvent showed that the starting material had been consumed. Figure 11 shows some of the possible fragmentation products 16–19. These might form by a light-catalysed fragmentation of the oxime N–O bond followed by a secondary reaction of the sulfonate radical such as hydrogen abstraction from the solvent (Figure 12). Termination of free radicals after irradiation could also occur by recombination which could give peroxide 22. This peroxide 22 would require heating, in a development step, or hydrolysis to release acid 16. In these studies, only evidence for 4-methylbenzenesulfonic acid 16 has been found. 1H NMR analysis of the crude product in D2O, from the irradiation of compounds 11 and 13, showed two strong aromatic doublets and an upfield singlet which matched the spectrum for the standard 4-methylbenzenesulfonic acid 16. This assignment was confirmed by comparison of the 13C NMR data with standards of 4-methylbenzenesulfonic acid 16 and 4-methylbenzenesulfinic acid 17. Again, the data matched those for the standard 16 including the chemical shift at 142.3 ppm of the quaternary carbon attached to the sulfur atom. This occurs at a different chemical shift of 150.5 ppm for the sulfinic acid 17. Compound 13 released acid more efficiently than compound 11 as the 1H NMR data were stronger and cleaner. It was difficult to identify other products from the crude mixtures. However, no nitriles such as compound 18 or 19, which might form from radical 20, were detected by 1H NMR in CD3OD or by the benzonitrile infrared (IR) stretch at 2228 cm−1. The fate of species 20 is unknown. A water extract of both products turned blue litmus red showing that acid-forming precursors were liberated in the photolysis. Light sensitivity is required for applications as photoacid generators making these compounds potentially useful in the field. However, they must also be soluble in appropriate solvents used in the industry such as propylene glycol methyl ether acetate (PGMEA) or ethyl lactate. Although compound 13 is soluble in these solvents, compound 11 has poor solubility in them.
Some possible products from the photochemical decomposition of compounds 11 and 13.
The proposed scheme for the fragmentation of oxime sulfonate 13 to release 4-methylbenzenesulfonic acid 16.
Conclusion
The crystal structures of compounds 11–14 verify their oxime stereochemistry. Photochemical decomposition of the representative compounds 11 and 13 gave 4-methylbenzenesulfonic acid 16 which was observed in the 1H and 13C NMR spectra of the crude product in D2O. Irradiation of both compounds 11 and 13 gave solutions in water that turned blue litmus paper red. This work provides evidence that the class of acid released from the irradiation of oxime sulfonates is a sulfonic acid, which might catalyse modification of a polymer resist during development.4,8,10,11 Irradiation of compound 11 did not give the expected 1,8-dicarbonitrile 18 and irradiation of compound 13 did not give benzonitrile 19 in easily detectable amounts. The efficient release of acid and good solubility suggest that compound 13 has potential use as a photoacid generator but acid is not liberated directly and requires a secondary hydrogen abstraction step or hydrolysis step.
Experimental
General: IR spectra were recorded on a diamond anvil spectrophotometer. UV spectra were recorded using a Perkin-Elmer Lambda 25 UV-Vis spectrometer with ethyl alcohol (EtOH) as the solvent. 1H and 13C NMR spectra were recorded at 600 and 150 MHz, respectively, using a Varian 400 spectrometer. Chemical shifts, δ, are given in ppm relative to the residual solvent and coupling constants, and J values are given in Hz. Low- and high-resolution mass spectra were obtained at the University of Wales, Swansea using electron impact ionisation and chemical ionisation. Melting point (m.p.) values were determined on a Kofler hot-stage microscope. Irradiations were done in a 100-mL immersion well with a Photochemical Reactors 6-W lamp (Reading, UK) and air cooling from a fume hood fan. No water flow was required with dichloromethane (DCM) as a solvent. Reflective foil was used to shield the lamp. The method is user friendly for students.
General procedure for di-oximes or mono-oximes
Acenaphthylene-1,2-dione di-oxime 9
A literature procedure was followed but the work-up was different.15 Acenaphthenequinone (5.0 g, 27.5 mmol), hydroxylamine hydrochloride (4.2 g, 60.4 mmol) and sodium acetate (5.0 g, 61 mmol) were stirred at room temperature (rt) in EtOH (150 mL) for 24 h. The mixture was gently refluxed for 2 h and then cooled. The mixture was poured into water (400 mL) and left to stand for 2 h as the product precipitated. This was filtered with a large sinter, washed with water (100 mL) and air dried to give the title compound (5.2 g, 98%) as an off-white solid, m.p. > 220 °C (from DCM/light petroleum ether 40–60). λmax (EtOH)/nm 325 (log ε 3.2), 232 (4.6) and 212 (4.5); νmax (Diamond) 3453w, 3018w, 2837w, 1489w, 1418w, 1347w, 1289w, 1228w, 1185w, 1146w, 1016m, 1000m, 937m, 854s, 825s, 773s, 611m, 539m and 443s; δH (600 MHz; CDCl3) 7.69 (2H, m), 7.97 (2H, d, J = 6.0) and 8.43 (2H, d, J = 6.0); δC (150 MHz; CDCl3) 125.5, 127.1, 127.8, 129.0, 130.6, 136.9 and 149.6; m/z (Orbitrap ASAP) 213.0659 (M+ + H, 100%) C12H9N2O2 requires 213.0659.
The bis-oxime of acenaphthenequinone 11 (400 mg, 1.9 mmol), 4-methylbenzenesulfonyl chloride (863 mg, 4.5 mmol) and Et3N (457 mg, 4.5 mmol) were stirred in CH2Cl2 (100 mL) for 24 h at rt. The clear organic layer was washed with water (100 mL × 2) and dried over MgSO4. The solution was concentrated under reduced pressure to a solid and then extracted three times by swirling with light petroleum ether (100 mL) which removed excess 4-methylbenzenesulfonyl chloride. Swirling with a smaller amount of DCM (30 mL) removed brown impurities and gave a product (470 mg, 48%). Proton NMR analysis showed this product to be impure, containing triethylammonium tosylate, so it was dissolved in DCM (300 mL) and extracted with water (100 mL × 3) and concentrated under reduced pressure to give the title compound (0.34 g, 35%) as a pale yellow solid, m.p. > 220 °C (from DCM/light petroleum ether 40–60). λmax (EtOH)/nm 333 (log ε 3.3), 316 (3.3), 245–280sh (3.5) and 229 (4.2); νmax (Diamond) 1596w, 1575w, 1490w, 1390w, 1368w, 1178s, 1093m, 816s, 773s, 685s, 661s, 615s and 458m; δH (600 MHz; CDCl3) 2.42 (6H, s), 7.31 (4H, d, J = 12.0), 7.62 (2H, t, J = 6.0 and 6.0), 7.92–7.95 (6H, m) and 8.33 (2H, d, J = 6.0); δC (150 MHz; CDCl3) 21.7, 126.7, 128.4, 128.8, 129.5, 129.7, 130.1, 130.5, 131.8, 138.8, 145.7, 155.2; m/z (Orbitrap ASAP) 521.0842 (M+ + H, 100%) C26H21N2O6S2 requires 521.0841; 179.0603 (naphthalene-1,8-dicarbonitrile + H, 50%) C12H7N2 requires 179.0609.
The mono-oxime of benzil 12 (2.0 g, 8.9 mmol),15 4-methylbenzenesulfonyl chloride (3.4 g, 17.8 mmol) and Et3N (2.1 g, 21.0 mmol) were stirred in CH2Cl2 (100 mL) for 24 h at rt.16 The clear organic layer was washed with water (100 mL × 2) and dried over MgSO4. The solution was concentrated under reduced pressure to an oil which was swirled with light petroleum ether (30 mL × 3) and left to crystallise. The solid was then extracted by swirling with light petroleum ether (30 mL × 7) and concentrated under reduced pressure. The solid was then dissolved in DCM (100 mL) and filtered through a pad of silica to give the title compound (1.8 g, 53%) as a pale yellow solid, m.p. 121 °C–122 °C (from DCM/light petroleum ether 40–60) on evaporation of the solvent. λmax (EtOH)/nm 256 (log ε 4.3), 232 (4.2) and 207 (4.6); νmax (Diamond) 1680s, 1594w, 1446w, 1371s, 1230m, 1174s, 1091w, 759s, 719s, 660s, 579s, 545s, 515s and 470m; δH (600 MHz; CDCl3) 2.38 (s, 3H), 7.37 (4H, m), 7.48 (3H, m), 7.57 (2H, d, J = 6.0), 7.66 (1H, t, J = 6.0 and 12.0) and 7.85 (4H, t, J = 6.0 and 6.0); δC (150 MHz; CDCl3) 21.8, 127.5, 128.9, 129.0, 129.2, 129.3, 129.5, 129.8, 132.2, 132.3, 133.7, 135.2, 145.5, 163.2 and 190.1; m/z (EI) 397.1211 (M+ + NH4, 100%) C21H21N2O4S requires 397.1217 (Orbitrap ASAP); 104. 0519 (benzonitrile + H, 100%) C7H6N requires 104.0500.
The mono-oxime of benzil 12 (1.0 g, 4.2 mmol), propanesulfonyl chloride (1.0 mL, 8.4 mmol) and Et3N (0.85 g, 8.4 mmol) were stirred in CH2Cl2 (100 mL) for 24 h at rt. The clear organic layer was washed with water (100 mL × 2) and dried over MgSO4. The solution was concentrated under reduced pressure to a solid and then extracted by swirling with light petroleum ether (30 mL × 10). This gave the title compound (1.3 g, 89%) as a colourless solid, m.p. 126 °C–127 °C (from DCM/light petroleum ether 40–60). λmax (EtOH)/nm 255 (log ε 3.7) and 208 (4.1); νmax (Diamond) 1680s, 1379m, 1367m, 1172s, 841m, 829m, 806s, 796s, 773s, 568s, 519m, 519m and 493m; δH (600 MHz; CDCl3) 1.09 (3H, t, J = 6.0 and 6.0), 1.92 (2H, m), 3.37 (2H, t, J = 6.0 and 8.0), 7.45 (2H, t, J = 6.0 and 12.0), 7.54 (3H, t, J = 6.0 and 12.0), 7.66–7.73 (3H, m) and 7.97 (2H, d, J = 12.0); δC (150 MHz; CDCl3) 12.8, 17.1, 51.2, 127.7, 128.8, 129.3, 129.4, 129.6, 132.5, 133.6, 135.4, 163.9 and 190.0; m/z (Orbitrap ASAP) 349.1215 (M+ + NH4, 20%). C17H21N2O4S requires 349.1222.
Photochemical irradiations
In total, 200 mg of compounds 11 or 13 were irradiated with a 6-W lamp for 5 h in 100 mL of CH2Cl2 without deoxygenation. The solution was concentrated and TLC analysis showed that extensive decomposition of the starting material had occurred. The crude products were both shown to contain 4-methylbenzenesulfonic acid 16 by 1H NMR. δH (400 MHz; D2O) 2.20 (3H, s), 7.11 (2H, d, J = 8.0) and 7.45 (2H, d, J = 8.0); δC (150 MHz; D2O) 20.4, 125.2, 129.3, 139.3 and 142.3. From the irradiation of compound 11, νmax (diamond anvil) 1678 cm−1; from the irradiation of compound 13, νmax (diamond anvil) 1682 cm−1. Standard of 4-methylbenzenesulfonic acid 16 δH (400 MHz; D2O) 2.10 (3H, s), 7.00 (2H, d, J = 8.0) and 7.41 (2H, d, J = 8.0); δC (150 MHz; D2O) 20.4, 125.2, 129.3, 139.3 and 142.3; standard of 4-methylbenzenesulfinic acid 17 δH (400 MHz; D2O) 2.29 (3H, s), 7.27 (2H, d, J = 8.0) and 7.46 (2H, d, J = 8.0); δC (150 MHz; D2O) 20.6, 123.5, 129.6, 141.1 and 150.5.
Crystal structure determinations
Single crystals of 11–14 were recrystallised from DCM/light petroleum ether solution. Intensity data for 11–14 were collected at T = 100 K using a Rigaku AFC11 charge-coupled device (CCD) diffractometer (Mo Kα radiation, λ = 0.71073 Å for 11 and 13 and Cu Kα radiation, λ = 1.54184 Å for 12 and 14). Each structure was easily solved by direct methods and the structural models were completed and optimised by least-squares refinement against |F|2 using SHELXL-2014.25 The crystal quality for 14 was notably poorer than for the other structures. For all structures, the H atoms were geometrically placed (C–H = 0.95–0.98 Å) and refined as riding atoms. The methyl groups were allowed to rotate, but not to tip, to best fit the electron density. The constraint Uiso(H) = 1.2Ueq(C) or 1.5Ueq(methyl C) was applied in all cases.
11: C26H20N2O6S2, Mr = 520.56, pale orange column, 0.23 × 0.06 × 0.05 mm3, triclinic, space group (No. 2), Z = 2, a = 7.9131 (2) Å, b = 11.5800 (3) Å, c = 14.4677 (3) Å, α = 103.676 (2)°, β = 95.088 (2)°, γ = 109.671 (2)°, V = 1192.28 (5) Å3 at 100 K. Number of measured and unique reflections = 20,472 and 5452, respectively (−10 ⩽ h ⩽ 10, −15 ⩽ k ⩽ 15, −18 ⩽ l ⩽ 18; 2θmax = 50.5°; RInt = 0.017). Final R(F) = 0.035, wR(F2) = 0.093 for 327 parameters and 4971 reflections with I > 2σ(I) (corresponding R-values based on all 5452 reflections = 0.039 and 0.095, respectively), Cambridge Crystallographic Data Centre (CCDC) deposition number 1870464.
12: C18H20N2O6S2, Mr = 424.48, pale yellow block, 0.23 × 0.21 × 0.18 mm3, triclinic, space group (No. 2), Z = 4, a = 7.06267 (6) Å, b = 11.08547 (10) Å, c = 25.1261 (2) Å, α = 96.2327 (6)°, β = 90.5797 (8)°, γ = 95.8034 (7)°, V = 1945.10 (3) Å3 at 100 K. Number of measured and unique reflections = 34,798 and 7043, respectively (−8 ⩽ h ⩽ 8, −13 ⩽ k ⩽ 13, −30 ⩽ l ⩽ 30; 2θmax = 136.5°; RInt = 0.015). Final R(F) = 0.029, wR(F2) = 0.082 for 509 parameters and 6850 reflections with I > 2σ(I) (corresponding R-values based on all 7043 reflections = 0.030 and 0.082, respectively), CCDC deposition number 1870465.
13: C21H17NO4S, Mr = 379.42, colourless block, 0.27 × 0.12 × 0.04 mm3, monoclinic, space group Ia (No. 9), Z = 4, a = 13.0319 (6) Å, b = 12.1421 (5) Å, c = 11.8439 (5) Å, β = 102.396 (5)°, V = 1830.42 (14) Å3 at 100 K. Number of measured and unique reflections = 10,604 and 3801, respectively (−16 ⩽ h ⩽ 15, −15 ⩽ k ⩽ 15, −15 ⩽ l ⩽ 15; 2θmax = 55.0°; RInt = 0.035). Final R(F) = 0.029, wR(F2) = 0.076 for 245 parameters and 3610 reflections with I > 2σ(I) (corresponding R-values based on all 3801 reflections = 0.031 and 0.077, respectively), Flack absolute structure parameter = 0.02 (4), CCDC deposition number 1870466.
14: C17H17NO4S, Mr = 331.37, colourless needle, 0.30 × 0.03 × 0.01 mm2, triclinic, space group (No. 2), Z = 2, a = 5.2869 (4) Å, b = 9.1995 (12) Å, c = 16.829 (2) Å, α = 97.497 (11)°, β = 95.822 (8)°, γ = 94.282 (9)°, V = 804.11 (16) Å3 at 100 K. Number of measured and unique reflections = 10,351 and 2857, respectively (−5 ⩽ h ⩽ 6, −10 ⩽ k ⩽ 10, −19 ⩽ l ⩽ 20; 2θmax = 135.0°; Rint = 0.129). Final R(F) = 0.119, wR(F2) = 0.307 for 209 parameters and 1982 reflections with I > 2σ(I) (corresponding R-values based on all 2857 reflections = 0.158 and 0.338, respectively), CCDC deposition number 1870467.
Footnotes
Acknowledgements
We are grateful to the National Mass Spectrometry Foundation, University of Swansea and to the National Crystallographic Service Centre, University of Southampton.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
CCDC deposition numbers 1870464–1870467 contain the supplemental crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre at
RobinsonAPGLawsonRA. Materials and processes for next generation lithography (Frontiers of Nanoscience, vol. 11) (ed. PalmerRE). Amsterdam: Elsevier, 2016.