
Abstract
Background:
Approximately one in four stroke patients suffer from recurrent vascular events, underlying the necessity to improve secondary stroke prevention strategies. Immune mechanisms are causally associated with coronary atherosclerosis. However, stroke is a heterogeneous disease and the relative contribution of inflammation across stroke mechanisms is not well understood. The optimal design of future randomized control trials (RCTs) of anti-inflammatory therapies to prevent recurrence after stroke must be informed by a clear understanding of the prognostic role of inflammation according to stroke subtype and individual patient factors.
Aim:
In this narrative review, we discuss (1) inflammatory pathways in the etiology of ischemic stroke subtypes; (2) the evidence on inflammatory markers and vascular recurrence after stroke; and (3) review RCT evidence of anti-inflammatory agents for vascular prevention.
Summary of review:
Experimental work, genetic epidemiological data, and plaque-imaging studies all implicate inflammation in atherosclerotic stroke. However, emerging evidence also suggests that inflammatory mechanisms are also important in other stroke mechanisms. Advanced neuroimaging techniques support the role of neuroinflammation in blood–brain barrier dysfunction in cerebral small vessel disease (cSVD). Systemic inflammatory processes also promote atrial cardiopathy, incident and recurrent atrial fibrillation (AF). Although several inflammatory markers have been associated with recurrence after stroke, interleukin-6 (IL-6) and high-sensitivity C-reactive protein (hsCRP) are presently the most promising markers to identify patients at increased vascular risk. Several RCTs have shown that anti-inflammatory therapies reduce vascular risk, including stroke, in coronary artery disease (CAD). Some, but not all of these trials, selected patients on the basis of elevated hsCRP. Although unproven after stroke, targeting inflammation to reduce recurrence is a compelling strategy and several RCTs are ongoing.
Conclusion:
Evidence points toward the importance of inflammation across multiple stroke etiologies and potential benefit of anti-inflammatory targets in secondary stroke prevention. Taking the heterogeneous stroke etiologies into account, the use of serum biomarkers could be useful to identify patients with residual inflammatory risk and perform biomarker-led patient selection for future RCTs.
Introduction
Secondary stroke prevention strategies have improved in recent decades. However, pooled data from contemporary studies demonstrate that the 5-year risk of major vascular recurrence after ischemic stroke or transient ischemic attack (TIA) ranges between 20% and 30%. 1 New targets for secondary prevention after stroke are needed to reduce this residual risk. Experimental work implicates immune mechanisms in atherosclerosis. 2 Recent evidence implicates inflammatory processes in the pathophysiology of atherosclerotic stroke, cerebral small vessel disease (cSVD) and atrial fibrillation (AF). Randomized control trials (RCTs) have demonstrated that anti-inflammatory therapies reduce the risk of vascular events, including stroke, in patients with coronary artery disease (CAD).3,4 For these reasons, inflammation is a promising therapeutic target for secondary stroke prevention. In this review, we will summarize the contribution of inflammation according to stroke etiology, review the prognostic information provided by inflammatory markers after stroke, and finally, discuss the potential use of anti-inflammatory therapies in clinical stroke care and their potential translation from the field of cardiovascular to cerebrovascular diseases. We will not focus on different cell lines relevant for the immune response but rather on some selected inflammatory signaling pathways and their respective biomarkers.
Methods
We performed a literature search using Ovid MEDLINE to identify studies evaluating inflammatory mechanisms in stroke recurrence and stroke etiology (Supplemental Material).
Large artery atherosclerotic stroke—role of inflammation
Large artery atherosclerosis (LAA) causing stenosis of the cervical and intracranial arteries is identified in 15–25% of ischemic strokes in Caucasians, with higher rates in Asian and Black patients. Non-stenosing atherosclerosis is also likely causal in a high proportion of cryptogenic strokes. In a pooled analysis of population-based studies, atherosclerotic stroke was associated with a higher risk of early recurrence (20% at 3 months) compared with other stroke etiologies. 5 Atherosclerosis is a chronic maladaptive inflammatory disorder. 2 Cardiovascular risk factors, such as hypertension or dyslipidemia, activate vessel endothelium promoting adhesion molecule expression which attracts leukocytes. 6 Monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8) promote leukocyte migration into the arterial intima, where mononuclear cells differentiate into macrophages, in turn engulfing lipid particles and becoming foam cells, a hallmark of early atherosclerosis.
The NLRP3 inflammasome is activated by oxidized low-density lipoprotein (LDL) within foam cells. 7 In turn, caspase-1 cleaves both pro-IL-1β and pro-IL-18 to their active forms. This process stimulates an inflammatory cascade of downstream expression of IL-6, tumor necrosis factor-alpha (TNF-α), other cytokines, and reactive oxygen species. 2 These inflammatory agents act locally within the developing plaque, recruiting T-lymphocytes and promoting the expression of tissue factor and matrix metalloproteinases (MMPs). Smooth muscle cells drive the formation of a fibrous cap of collagen and fibrin covering the lipid core. Failure in the usual homeostatic-apoptotic process results in constant renewal of the local inflammatory cascade: IL-1β and TNF-α stimulate overexpression of MMPs, which erode the fibrous cap. Cap rupture exposes the necrotic core containing tissue factor, triggering a clotting cascade and coronary or cerebral thromboembolism. Atherosclerotic plaque stability relies on the delicate balance between proinflammatory and repair mechanisms. Modulation of these inflammatory processes within atherosclerotic plaques is an attractive therapeutic target in atherosclerotic stroke but also irrespective of the most likely cause of the index stroke, as the focus would be to prevent stroke recurrence.
In addition, brain infarction itself appears to accelerate atherosclerosis. Data from animal models demonstrate that cell-free DNA 8 and other soluble mediators 9 released from recently ischemic brain promote a systemic immune response. In apolipoprotein-deficient mice, which develop premature atherosclerosis, this post-stroke immune response accelerates existing plaque development and increases recurrent stroke risk. Moreover, interruption of this post-stroke inflammatory cascade with DNAse or inflammasome inhibition can reduce atherosclerotic lesion development and ameliorate recurrence risk. 8 Stroke is strongly associated with age and the impact of immune cell aging—also called immunosenescence—on atherosclerosis is highlighted in recent literature. Immunosenescence describes an age-dependent decline of immune cell functioning leading to a secretion of proinflammatory mediators further driving atherosclerotic plaque formation. 10
Data from clinical studies link inflammation to LAA stroke. The severity of immune cell infiltration in resected carotid plaque is associated with morphological evidence of plaque instability 11 and stroke recurrence. 12 In patients with recently symptomatic carotid stenosis, plaque inflammation imaged by positron emission tomography (PET) is strongly associated with early- and late-outcome recurrent stroke. 13
Small vessel stroke—role of inflammation
cSVD represents a major cause of hemorrhagic stroke and causes approximately 20% of ischemic strokes. 14 In addition, cSVD can elicit vascular dementia 15 and gait dysfunction, 16 and thus has a major impact on the global health of older people.
Despite the clinical importance of cSVD, the underlying pathophysiology is not entirely understood. Recent studies hypothesized that blood–brain barrier permeability and neuroinflammation are causal to SVD development and progression.17,18 This hypothesis is supported by experimental data from rodent models. 19 Jalal et al. described blood barrier disruption in rats after induction of hypoxia followed by an MMP-9-mediated infiltration of immune cells located around the affected small vessels. Advanced neuroimaging techniques provide relevant insight into potential pathophysiological processes in cSVD including the role of neuroinflammation. In one small study that used PET–magnetic resonance imaging (MRI), blood–brain barrier permeability and microglial activation were both increased in patients with sporadic cSVD, but appeared to be spatially distinct processes. 17
In addition, inflammatory markers, such as C-reactive protein (CRP) and IL-6, have been associated with the presence 20 of SVD and CRP was associated with the progression of cSVD 21 in stroke-free subjects. In a substudy of the SPS3 trial, circulating high-sensitivity C-reactive protein (hsCRP) was independently associated with recurrent stroke after lacunar infarction. 22
Cardioembolic stroke—role of inflammation
Multiple recognized risk factors are related to cardioembolic stroke 23 and about one-third of ischemic strokes are attributed to cardioembolism. 14 Several lines of evidence support the role of inflammation in the pathogenesis of AF. Systemic inflammatory triggers secondary to obesity, hypertension, and viral infection may promote the development of AF. 24 CRP is associated with incident AF in population-based studies 25 and incident stroke in patients with established AF. 26 Elevated IL-6 levels have also been attributed to a twofold increased risk of stroke in patients with AF. 27 In addition, a gradual decrease in systemic CRP levels was reported after restoration of sinus rhythm in AF, suggesting that systemic inflammation is associated with AF. 28 Moreover, the NLRP3 inflammasome is hypothesized to be an important contributor to the occurrence and maintenance of AF. 27 Oxidative stress can lead to a rise of intracellular calcium 29 which activates the NLRP3 inflammasome. 30 The NLRP3 inflammasome is also upregulated in cardiomyocytes in patients with AF. 31 Finally, anti-inflammatory therapies may reduce AF burden and this may have important implications for prevention trials after cardioembolic stroke. Colchicine was associated with a non-significant trend toward reduced incident AF in a pooled analysis of two RCTs of colchicine therapy in CAD. 32
Inflammatory blood biomarkers and ischemic stroke
HsCRP
CRP is a nonspecific marker of inflammation, synthesized in the liver in response to upstream IL-6 signaling. In a pooled sample of 160,309 adults, CRP was associated with first-ever stroke independent of cardiovascular risk factors. 33 American guidelines recommend hsCRP measurement for risk-stratifying patients for primary cardiovascular disease prevention in patients at intermediate risk. 34 Until recently, there was conflicting data on the association between CRP and recurrence after stroke. However, leveraging individual participant data (IPD), the Blood Inflammatory markers in Stroke Collaboration (BISC) reported that hsCRP was independently associated with major adverse cardiovascular events (MACE) in 8420 patients with ischemic stroke/TIA after adjustment for cardiovascular risk factors and secondary prevention therapy. 35 CRP is an attractive biomarker due to its stability in whole blood and frozen stored samples, availability of standardized assays, and ease of measurement. However, despite its potential utility as a risk marker, Mendelian randomization studies suggest that CRP is unlikely to be causal in the pathogenesis of atherosclerosis. 36 Therefore, associations with stroke recurrence probably reflect upstream IL-1β–IL-6 axis signaling.
IL-6
A genome-wide association study (GWAS) of 200,000 individuals demonstrated that downregulated IL-6 signaling (analogous to pharmacological blockade of IL-6 receptor) was associated with an 11% reduction in risk of ischemic stroke, with larger effect sizes (29% reduction) for large artery and small vessel stroke, but no association with cardioembolic stroke. 37 In people without a history of vascular disease, circulating IL-6 is independently associated with first stroke. 38 Elevated IL-6 predicts carotid atheroma plaque severity, vulnerability, and progression. 39 Initial studies reported conflicting findings regarding the association between circulating IL-6 and post-stroke recurrence. 40 However, in IPD data from BISC, IL-6 was again independently associated with MACE and recurrent stroke irrespective of timing of phlebotomy or stroke severity. 35
MCP-1
MCP-1 (also referred to as chemokine (C-C motif) ligand-2 (CCL2)) and its receptor (CCR2) play an important role in early plaque development, by facilitating monocyte intimal recruitment in response to proatherogenic stimuli, including LDL. 41 MCP-1 levels are associated with histopathological, clinical, and molecular hallmarks of plaque vulnerability. 42 In a meta-analysis of over 17,000 stroke-free adults with 16 years of follow-up, elevated MCP-1 was associated with stroke, independent of cardiovascular risk factors, IL-6, and CRP. 43 Genetic epidemiological data have demonstrated that MCP-1 is independently associated with a higher risk of stroke, particularly for large artery or cardioembolic stroke. 44 However, data from stroke cohorts are lacking and limited to just two studies, neither of which demonstrated associations with recurrent stroke.45,46 The reasons for these conflicting findings are uncertain, but may relate to low statistical power, over-representation of lacunar stroke in the study population, or short follow-up. While MCP-1 is a promising target for reducing atherosclerotic plaque vulnerability, further data are needed in patients with stroke before embarking on RCTs of agents targeting the CCL2–CCR2 axis.
TNF-α
TNF-α is a potent proinflammatory cytokine. Its receptors are widely expressed in multiple cell types, facilitating activation of a wide range of inflammatory cytokines. It contributes to the development of atherosclerosis by increasing endothelial permeability to other inflammatory cells, monocytes, and lipid particles and activating MMP-9 production. Mendelian randomization analysis in 30,000 individuals without cardiovascular disease demonstrated that genetically predicted TNF-α levels were associated with an increased risk of ischemic stroke and CAD. 47 In a meta-analysis of 29 population studies, TNF-α levels were also associated with an increased risk of myocardial infarction (MI) and cardiovascular death in a dose-dependent manner. 48 However, in a case-control study nested in an RCT, TNF-α was not associated with ischemic stroke in older adults. By contrast, in PROGRESS, raised TNF-α was associated with increased risk of recurrent stroke and was the only inflammatory protein to retain this association after extensive adjustment for confounding risk factors. 49 Further studies are needed to strengthen the rationale for targeting TNF-α for secondary prevention therapies after stroke.
Inflammatory markers in clinical practice
Several issues warrant further research before measurement of inflammatory biomarkers to improve clinical risk prediction in routine practice or to select patients for anti-inflammatory therapies in RCTs. First, further studies are needed to clarify the role of intra-individual variation of inflammatory markers on future risk prediction after stroke. Second, the effect of anti-inflammatory therapies on inflammatory markers is incompletely understood. Third, the timing of blood draw in relation to the infarct may influence biomarker concentrations. Although data from proteomic studies indicate a generalized peripheral immune response to stroke, many inflammatory signatures remain significantly elevated for months to years after stroke. 50 Further studies are needed to inform clinical interpretation of raised inflammatory markers after stroke.
Further information on the role of other inflammatory markers (IL-8, IL-18, MMP-8, CXCL16, CD40, tissue factor) and stroke risk are discussed in the Supplemental Material.
Clonal hematopoiesis of indeterminate potential
Clonal hematopoiesis of indeterminate potential (CHIP) is the presence of a clonally expanded hematopoietic stem cell caused by a leukemogenic mutation in individuals without evidence of hematologic malignancy, dysplasia, or cytopenia, and is associated with a 1% risk of leukemia per annum. 51 The prevalence of CHIP increases with age due to the accumulation of mutational processes over time. 52 CHIP is independently associated with a twofold increased risk of cardiovascular events. 52 CHIP is believed to enhance vascular risk by priming monocytes to release excessive inflammatory cytokines. Mouse models of CHIP demonstrate accelerated atherosclerotic lesion formation and widespread upregulation of inflammatory proteins. 53 CHIP is also associated with increased risk of first stroke. 54 In a prospective cohort study of stroke patients, 41% had somatic mutations, and the presence of CHIP was associated with LAA stroke and white matter lesions. Patients with CHIP had markedly higher proinflammatory proteins, including IL-6 and hsCRP, and were at higher risk of recurrent vascular events. Intriguingly, the association was partially mitigated by the presence of a common variant of the IL-6 receptor, which is known to impair IL-6 signaling, suggesting that these somatic mutations may at least partially mediate their proatherogenic effects through enhanced IL-6–CRP axis stimulation. 55 Others have reported that the presence of atherosclerosis may accelerate clonal hematopoiesis. 56 These data link the accumulation of somatic mutations in myeloid lines with an increased residual vascular risk, which appear to be driven an interacting cycle of inflammation and atherosclerosis.
Translation to clinical practice
Anti-inflammatory therapies for vascular prevention after stroke
There are several anti-inflammatory and immunomodulatory trials in the context of acute stroke treatment to prevent unfavorable functional outcome, but there are no published RCTs of anti-inflammatory therapies for secondary prevention after stroke. All completed RCTs of anti-inflammatory therapy for secondary prevention derive from the coronary literature. In this section, we will discuss the evolution of anti-inflammatory therapies and their potential application in stroke (also see Figure 1).
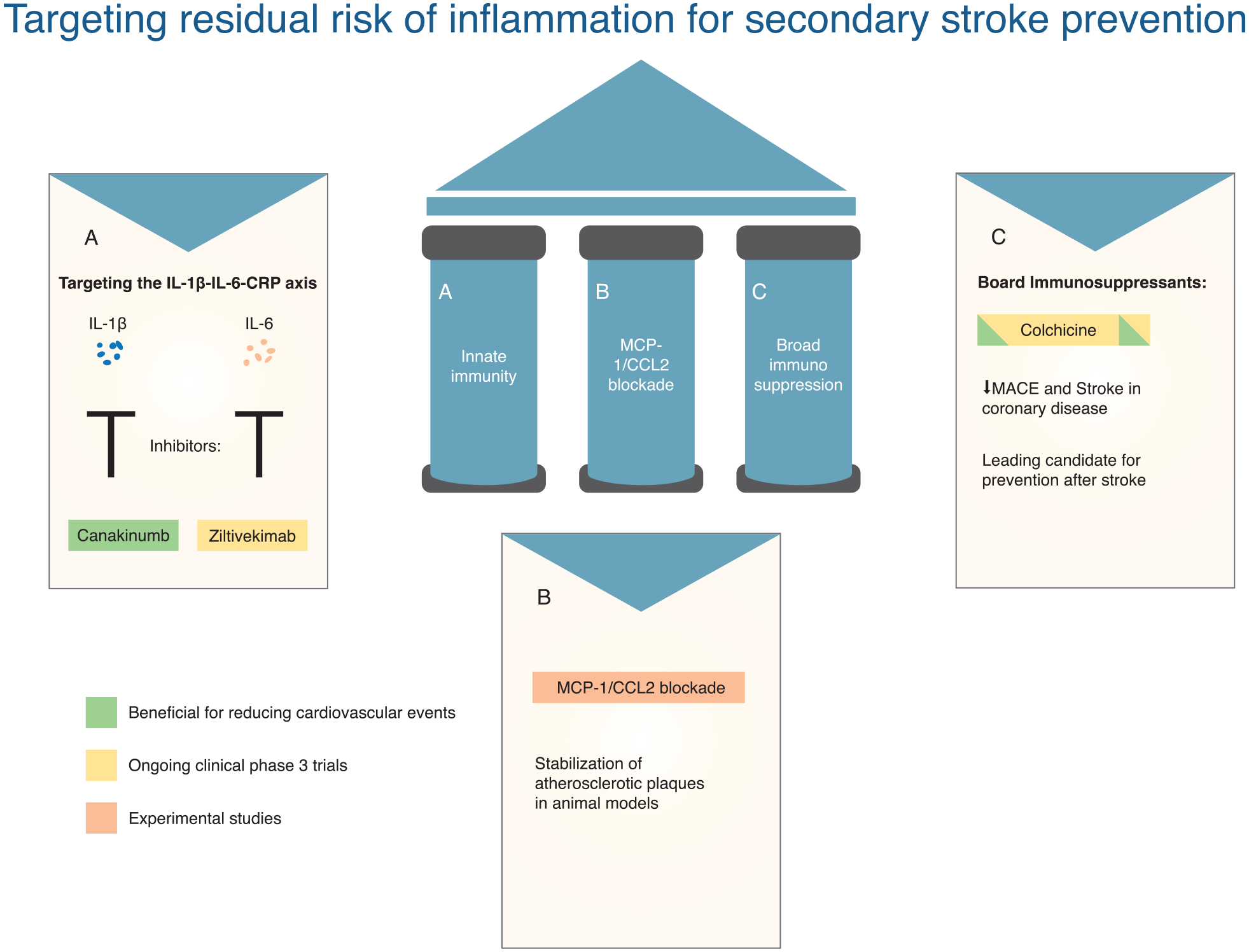
Targeting residual risk of inflammation for secondary stroke prevention.
Residual inflammatory risk
Experimental and clinical data support pleiotropic effects of statins beyond lipid-lowering, including anti-inflammatory effects. Over 20 years ago, data from the AFCAPS/Tex-CAPS trials indicated an interaction between the treatment effect of statins and baseline CRP–lipid levels. A protective effect of lovastatin was observed for coronary events in participants with elevated CRP despite a sub-median LDL. By contrast, statin therapy had no effect in patients with both low CRP and low LDL. 57 The Women’s Health Study also showed an incremental increase in the risk of MACE across subgroups defined by baseline CRP–LDL levels. 58 More recently, a pooled analysis of 31,000 high-risk patients receiving contemporary statins from three RCTs showed that the risk of MACE was significantly increased in patients with elevated baseline hsCRP ⩾ 2 mg/L irrespective of baseline LDL levels. The risk of vascular events was not increased in patients with residual cholesterol risk (RCR) (hsCRP < 2 mg/L and LDL ⩾ 1.8 mmol/L). In contrast, patients with residual inflammatory risk (RIR) (hsCRP ⩾ 2 mg/L and LDL < 1.8 mmol/L) had an 18% increased risk of MACE. 59 In the SPIRE trial, which evaluated proprotein convertase subtilisin/kexin type 9 (PCSK-9) inhibitors, patients with hsCRP ⩾ 3 mg/L had greater than 60% increased risk of MACE compared with patients with baseline hsCRP ⩽ 1 mg/L despite median LDL levels of 1.06 mmol/L. 60
Similar work has reflected a near-identical pattern of RIR in stroke patients. In a large prospective study in China, patients with RIR had a 30% increased risk of recurrent stroke, which was attenuated after adjustment for cardiovascular risk factors and secondary prevention. However, there was no increased risk in patients with RCR and risk was highest in patients with residual cholesterol inflammatory risk (RCIR) (LDL ⩾ 1.8 mmol/L, hsCRP > 3 mg/L). 61 In absolute terms, the risk of 1-year recurrent stroke was 13.1% in patients with RCIR and 8.4% without RCIR. These data support the hypothesis that a combined strategy of aggressive LDL and hsCRP-lowering in patients with stroke/TIA may yield added benefits compared with pursuing either strategy alone. Such an approach could be tested following stroke in an RCT with a 2 × 2 factorial design, with PCSK-9 inhibitors and anti-inflammatory therapy compared with best medical therapy (BMT).
Colchicine in cardiovascular diseases
Colchicine is an anti-inflammatory which inhibits microtubule and inflammasome assembly, reduces cytokine/adhesion molecule expression, and reduces inflammatory cell mitosis and motility. 62 In a substudy of an RCT, colchicine reduced on-treatment levels of hsCRP by 0.5 mg/L and reduced several proteins downstream of the NLRP3 inflammasome, including IL-6 and IL-1β. However, the anti-inflammatory effects of colchicine also extended to the adaptive immune system, hemostasis, and neutrophil degranulation. 63
In 2013, Colchicine was first evaluated for secondary prevention in a phase-3 prospective, randomized, observer-blinded endpoint (PROBE) designed trial (LoDoCo) in patients with stable CAD. Low-dose colchicine lowered the risk of 3-year MACE by two-thirds with a number needed to treat (NNT) of 11 compared with BMT. 64 The findings were confirmed in LoDoCo2, which reported a 30% reduced risk of MACE in stable coronary patients treated with colchicine. 65 The COLCOT trial, which recruited patients with recent MI, reported similar results. 66 In a subsequent meta-analysis of four RCTs (Supplemental Material), colchicine reduced the risk of MACE by 26%, with supportive direction of effect for MI and revascularization, but not cardiovascular death. The risk of stroke with colchicine was halved. 3 In the LoDoCo2 trial, there was a non-statistically significant increased risk of non-cardiovascular death in the colchicine arm (excess of 0.2 events per 100 person-years). 65 In the meta-analysis of the four trials, a similar trend toward an increased risk of non-cardiovascular death was shown, but the results were largely driven by the findings in LoDoCo2 and were not explained by an increased risk of infection, pneumonia, or cancer. Colchicine was well tolerated and not associated with the increased risk of gastrointestinal side effects or myalgias. 3 The findings of these trials led to a change of clinical practice recommendation in the recent guidelines of the European Society of Cardiology and colchicine was approved for treatment of cardiovascular disease by the FDA in June 2023. 67 Advantages of colchicine include low cost, wide availability globally including in low- and middle-income countries, good tolerability, and established safety profile at low doses. However, colchicine is contraindicated in patients with severe chronic kidney disease (CKD) and the known drug interactions should be taken into consideration.
Colchicine in secondary stroke prevention
Colchicine is now under evaluation for the secondary prevention of vascular events after stroke in the CONVINCE trial (NCT02898610), with results expected in 2024. In total, 3154 non-cardioembolic stroke patients were enrolled. Several other RCTs evaluating the effect of colchicine after stroke are either underway or are planned. CHANCE-3 (NCT05439356) is a placebo-controlled double-blind RCT currently recruiting patients with acute non-cardioembolic minor stroke or TIA with elevated hsCRP ⩾ 2 mg/L in China. The primary outcome is recurrent stroke at 90 days and results are anticipated in 2025. The RIISC-THETIS trial (NCT05476991) has a 2 × 2 factorial design and will evaluate the efficacy of ticagrelor and colchicine compared with aspirin in patients with atherosclerotic stroke.
Targeting the IL-1β–IL-6–CRP axis
Canakinumab is a human monoclonal antibody (mAb) to IL-1β, interrupts the central inflammatory signaling pathway, and is associated with substantial reductions in circulating levels of hsCRP–IL-6. The CANTOS trial enrolled participants with a history of MI and a hsCRP of > 2 mg/L despite aggressive secondary prevention therapy and showed that canakinumab reduces the risk of MACE. However, canakinumab was associated with a higher risk of death due to sepsis/infection (excess of 0.13 deaths per 100 person-years), neutropenia, and thrombocytopenia. 4 A greater magnitude of benefit was observed for MACE in patients achieving IL-6 suppression on canakinumab, but no benefit was seen in patients with persistently elevated IL-6. A marked reduction on cardiovascular and all-cause mortality was also observed in patients achieving IL-6 suppression. 68 A similar pattern was demonstrated in participants who achieved on-treatment hsCRP levels of < 2 mg/L. 69 The CANTOS trial demonstrated proof-of-principle that targeting the IL-6–CRP axis can reduce MACE in patients with established atherosclerosis and benefit was proportionate to the extent of IL-6–CRP inhibition.
Ziltivekimab is an mAb IL-6 inhibitor which binds directly to the IL-6 ligand and administered by monthly subcutaneous injections. In a phase-2 trial, which recruited patients with CKD and hsCRP ⩾ 2 mg/L, ziltivekimab lowered circulating hsCRP levels by up to 92% without any serious adverse events (AEs) and had a neutral effect on lipids. 70 The ZEUS trial is a phase-3 RCT which is recruiting patients with CKD, atherosclerotic cardiovascular disease risk, and with elevated hsCRP. Participants are randomized to subcutaneous ziltivekimab versus placebo and the primary outcome is any MACE during follow-up (NCT05021835).
Other potential therapeutic targets
There are no randomized data for TNF-α blockade in stroke. Observational data suggested that methotrexate use in patients with rheumatoid arthritis (RA) was associated with a reduced risk of vascular events. 71 However, a trial which randomized patients with CAD and diabetes/metabolic syndrome to methotrexate or placebo, demonstrated no benefit for vascular recurrence. 72
MCP-1–CCL2 blockade in mice has been shown to stabilize atherosclerotic plaques by reducing plaque macrophage content, neointimal hyperplasia, and inducing plaque fibrosis. 73 Apart from two small phase-2 studies, there are no ongoing trials with “hard” clinical endpoints in stroke or coronary disease. Nevertheless, epidemiological data supporting the importance of MCP-1 in atherosclerosis suggest that pharmacological manipulation of this pathway has potential clinical value. Drug design is challenging due to complexity of the chemokine ligand–receptor network, and complexity of the molecular structure of MCP-1 and its receptor. 74
Conclusion and future directions
Data from animal models not only have been highly informative in developing a conceptual framework for understanding immune mechanisms in stroke and atherosclerosis but also have limitations in drug discovery in human disease. Biomarker research and Mendelian randomization studies have provided important insights for therapeutic targets in atherosclerosis. Stroke is a heterogeneous disease with variable contribution of inflammation across stroke mechanisms. The potential benefit of anti-inflammatory therapies within individual patients with stroke may also vary. Consequently, unselected enrollment of patients in RCTs of such agents may dilute their potential efficacy in certain patient subgroups or stroke subtypes. A subtype-specific and biomarker-led approach to patient selection in future RCTs may improve the likelihood of detecting therapeutic benefit. However, more data are needed on the association of inflammatory markers with recurrence according to stroke mechanism and absolute biomarker level thresholds to define risk. Nevertheless, targeting inflammation to reduce vascular recurrence after stroke is highly promising and more RCTs of anti-inflammatory therapies for secondary prevention are needed.
Supplemental Material
sj-docx-1-wso-10.1177_17474930231207777 – Supplemental material for Targeting inflammation to reduce recurrent stroke
Supplemental material, sj-docx-1-wso-10.1177_17474930231207777 for Targeting inflammation to reduce recurrent stroke by Annaelle Zietz, Sarah Gorey, Peter J Kelly, Mira Katan and John J McCabe in International Journal of Stroke
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.Z., S.G., and J.J.M. report no conflict of interest. P.J.K. is the principal investigator of the CONVINCE trial. M.K. reports nonfinancial support from Roche and BRAHMS Thermo Fisher Scientific and grants from Swiss National Science Foundation.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: P.J.K. is funded by awards from the Health Research Board Ireland and Irish Heart Foundation for CONVINCE and research on biomarkers. M.K. received funding from the Swiss National Science Foundation, Swiss Heart Foundation, and Baasch-Medicus Foundation.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.