
Abstract
Background
The recommended maximum age and time window for intravenous alteplase treatment of acute ischemic stroke differs between the Europe Union and United States.
Aims
We compared the effects of alteplase in cohorts defined by the current Europe Union or United States marketing approval labels, and by hypothetical revisions of the labels that would remove the Europe Union upper age limit or extend the United States treatment time window to 4.5 h.
Methods
We assessed outcomes in an individual-patient-data meta-analysis of eight randomized trials of intravenous alteplase (0.9 mg/kg) versus control for acute ischemic stroke. Outcomes included: excellent outcome (modified Rankin score 0–1) at 3–6 months, the distribution of modified Rankin score, symptomatic intracerebral hemorrhage, and 90-day mortality.
Results
Alteplase increased the odds of modified Rankin score 0–1 among 2449/6136 (40%) patients who met the current European Union label and 3491 (57%) patients who met the age-revised label (odds ratio 1.42, 95% CI 1.21−1.68 and 1.43, 1.23−1.65, respectively), but not in those outside the age-revised label (1.06, 0.90−1.26). By 90 days, there was no increased mortality in the current and age-revised cohorts (hazard ratios 0.98, 95% CI 0.76−1.25 and 1.01, 0.86–1.19, respectively) but mortality remained higher outside the age-revised label (1.19, 0.99–1.42). Similarly, alteplase increased the odds of modified Rankin score 0-1 among 1174/6136 (19%) patients who met the current US approval and 3326 (54%) who met a 4.5-h revised approval (odds ratio 1.55, 1.19−2.01 and 1.37, 1.17−1.59, respectively), but not for those outside the 4.5-h revised approval (1.14, 0.97−1.34). By 90 days, no increased mortality remained for the current and 4.5-h revised label cohorts (hazard ratios 0.99, 0.77−1.26 and 1.02, 0.87–1.20, respectively) but mortality remained higher outside the 4.5-h revised approval (1.17, 0.98–1.41).
Conclusions
An age-revised European Union label or 4.5-h-revised United States label would each increase the number of patients deriving net benefit from alteplase by 90 days after acute ischemic stroke, without excess mortality.
Keywords
Introduction
Contraindications to alteplase treatment for stroke in FDA and EMA approved product inserts and AHA and ESO guidelines.
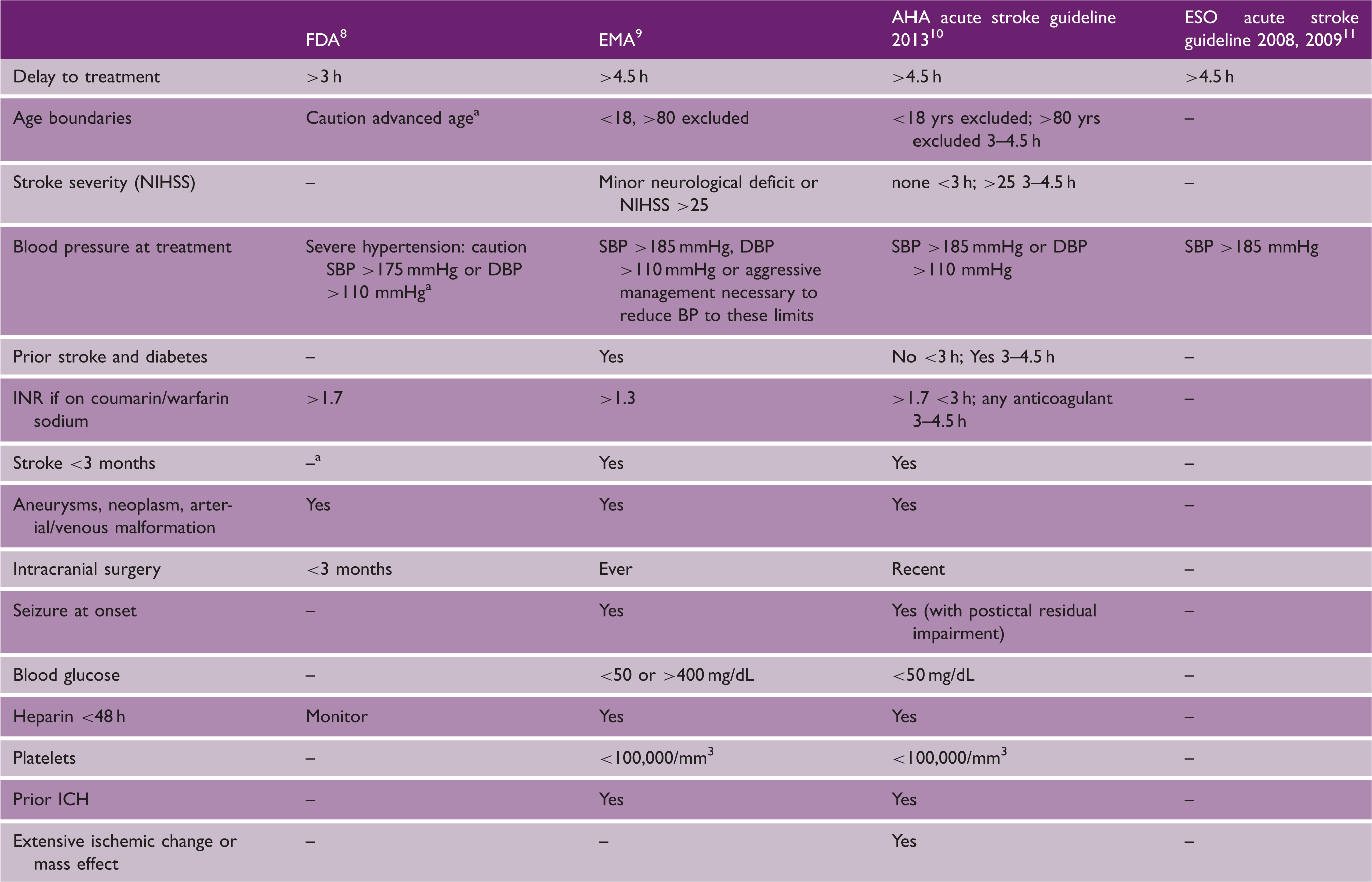
AHA: American Heart Association; DBP: diastolic blood pressure; EMA: European Medicines Agency; ESO: European Stroke Organisation; FDA: Food and Drug Administration; SBP: systolic blood pressure; ICH: Intracerebral haemorrhage; NIHSS: National Institutes of Health Stroke Scale.
States “the risks of bleeding are increased and should be weighed against the benefits”, not contraindication.
We have previously shown that intravenous alteplase increases the odds of achieving an excellent outcome (modified Rankin score (mRS) 0–1), and of gaining an improvement in mRS when used within 4.5 h, with earlier treatment yielding a larger benefit, and that the benefit is independent of age.12,13 While achieving this functional benefit, alteplase also increases the early risk of fatal intracerebral hemorrhage but, if treatment is given within 4.5 h, this is completely offset by a later survival benefit. Hence, by 90 days, there was no excess risk of mortality.12,14 Indeed, long-term survival data from the largest trial of alteplase in acute ischemic stroke, the third international stroke trial, suggest that further gains in survival might continue to accrue among early treated patients beyond 90 days. 15
Aims
Using the individual data available in the Stroke Thrombolysis Trialists’ Collaboration (STT), we sought to determine the likely impact on estimates of the benefits and potential harm of alteplase if current US and EU market approvals were modified to recommend treatment up to 4.5 h with no upper age restriction.
Methods
Patient-level data from nine randomized trials of alteplase in acute ischemic stroke were available,12,16–22 but one of those trials (ECASS I 17 ) was excluded from the current analysis because it tested alteplase at 1.1 mg/kg rather than the approved dose of 0.9 mg/kg. For the eight other trials that tested alteplase at a dose of 0.9 mg/kg, the search and statistical methods and outcomes used in the current report are the same as those used in previous publications,12–14,23 with the exception that analyses were conducted separately in respect of the EU and US labels. For each label, we aimed to examine the effects of alteplase among: (1) those who would have met existing criteria; (2) those who would have met hypothetical revisions (i.e. no upper age limit in the EU or a 4.5-h time window in US), and (3) those who would have remained outside revised labels.
In brief, for each of these sets of patients, the proportional effects of alteplase on an excellent stroke outcome (mRS 0–1), on symptomatic intracerebral hemorrhage (sICH, defined in three ways: type 2 parenchymal hemorrhage (PH-2) within seven days; Safe Implementation of Thrombolysis in Stroke Monitoring Study’s (SITS-MOST) hemorrhage within 24–36 h and on fatal intracerebral hemorrhage within seven days) were estimated by trial-stratified logistic regression. For subgroup analyses by treatment duration (≤3·0 h, >3 to ≤4·5 h, and >4·5 h), age (≤80 years and >80 years), and stroke severity (NIHSS ≤4, 5–10, 11–15, 16–21, and ≥22), these estimates were further adjusted for these three baseline features and for the relevant interaction term(s). The proportional effect of alteplase on 90-day mortality, overall and within the previously presented 12 risk periods 1–7 days, 8–30 days, and 31–90 days, was estimated using trial-stratified Cox regression without adjustment for other characteristics, while estimates of the common odds ratio for any improvement in mRS with alteplase were obtained for each trial using a proportional odds model adjusted for treatment allocation, with results subsequently meta-analyzed across trials.24,25 For each set of patients, sensitivity analyses re-estimated these main effects after additional adjustment for baseline treatment delay, age, and stroke severity.
In seven of the eight trials, mRS was assessed at three months, but in the third international stroke trial (IST-3) the Oxford Handicap Scale, which is similar to mRS, was assessed at six months. As pre-specified 23 we present functional outcome (mRS) at 3–6 months, but mortality at 90 days.
All estimates of treatment effect are provided with their 95% CIs with p values that are deemed conventionally significant, without allowance for multiple testing, at the 5% significance level. Analyses were done using SAS version 9.3 (SAS Institute, Cary) and R version 2.11.1 (https://www.R-project.org).
Results
Effects among patients who would have met the current EU label
Baseline characteristics of participants involved in the eight trials that tested alteplase 0.9 mg/kg versus control, subdivided according to whether the participant met all of the criteria in the current EU or US labels
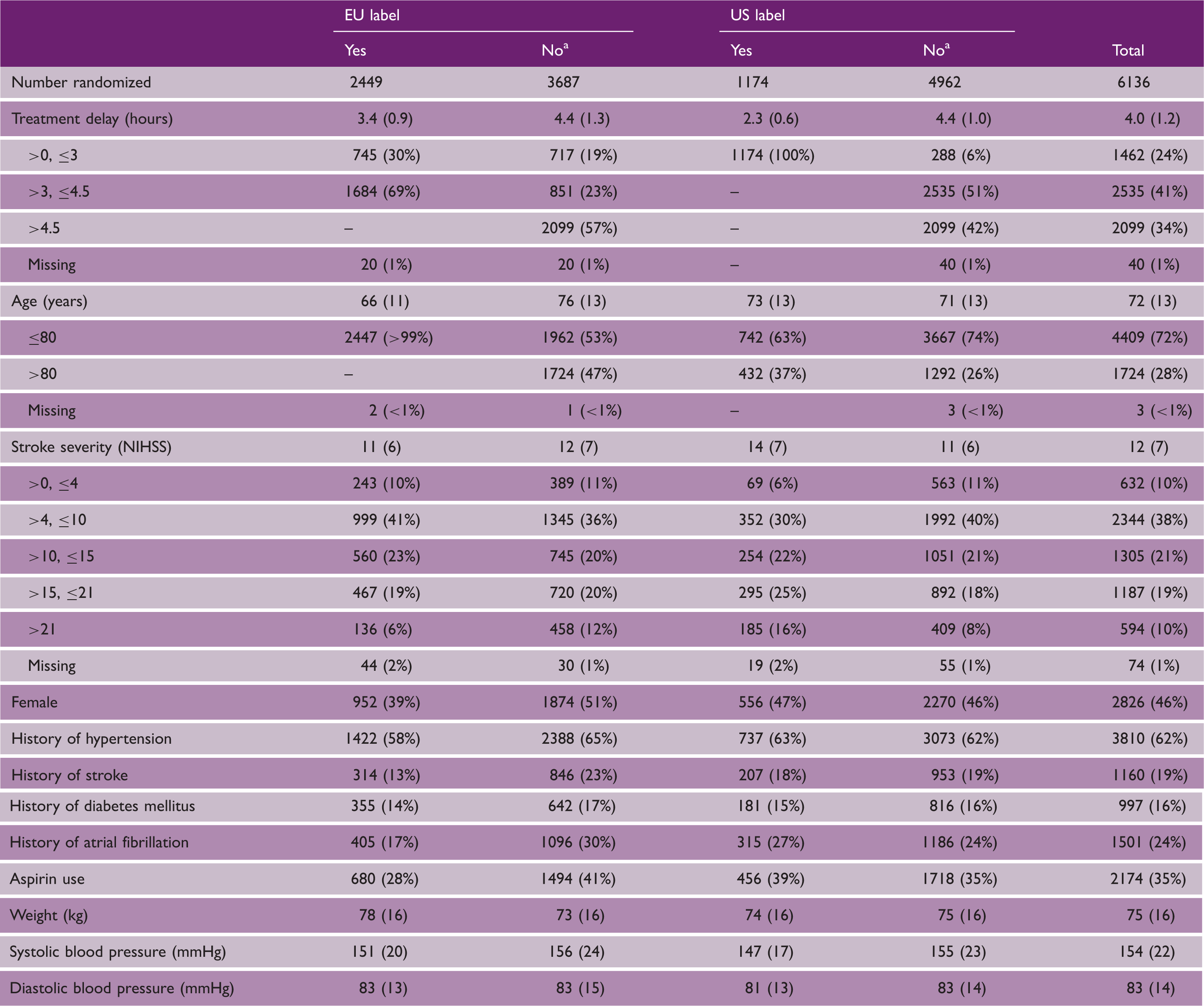
Categorical data presented as n (column %), continuous data presented as mean (SD).
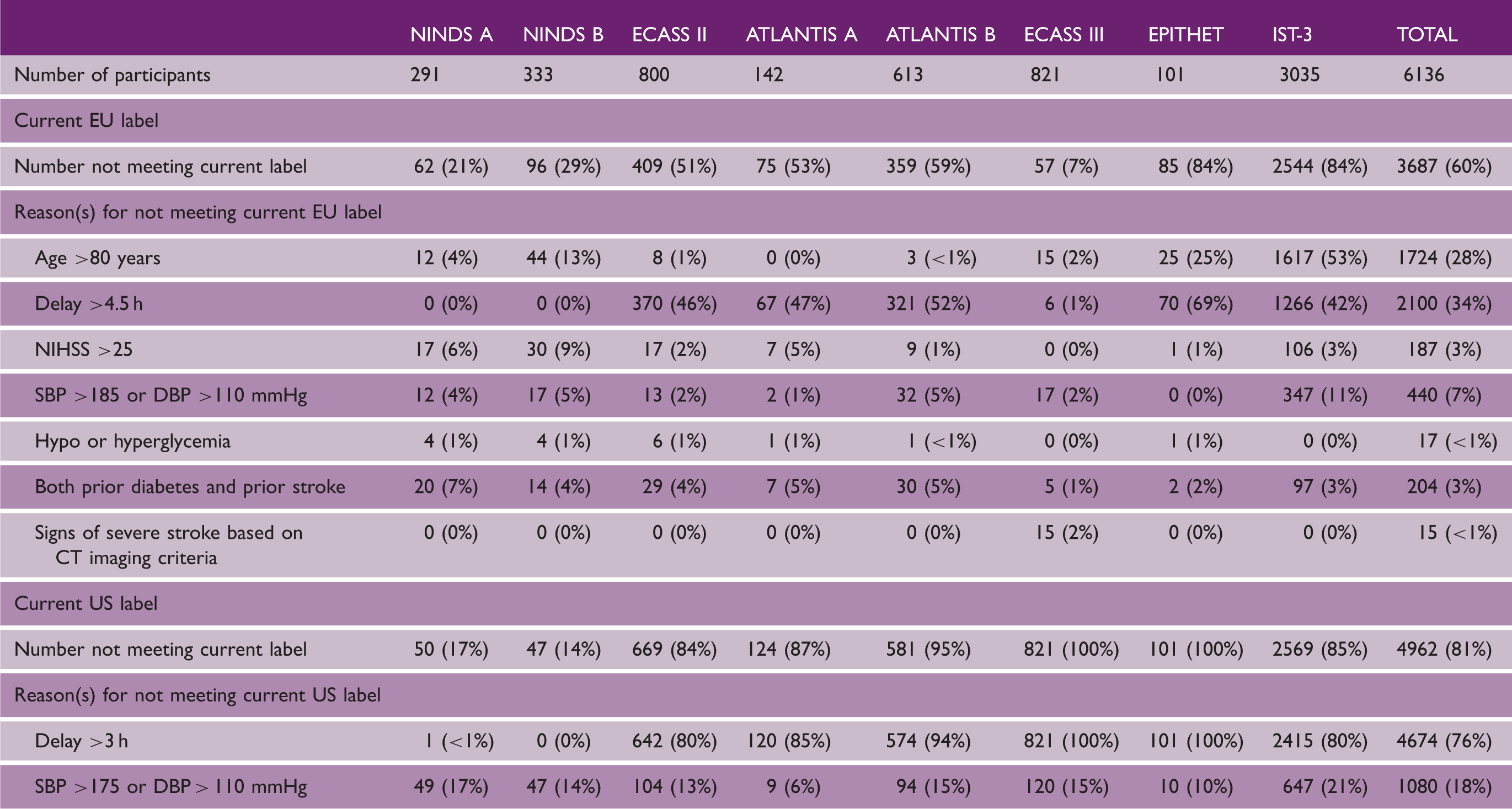
Participants can fail to meet the current label for any of several possible reasons (listed in Table 3). For example, of the 1568 participants who failed to meet the current EU label despite receiving treatment within 4.5 h, 1270 (81%) failed because they were aged >80 years and the remaining 298 (19%) failed for other reasons, while of the 288 patients who failed to meet the current US label despite receiving treatment within 3 h, all either had SBP >175 or DBP >110 mmHg.
Reasons for not meeting the current EU and US labels
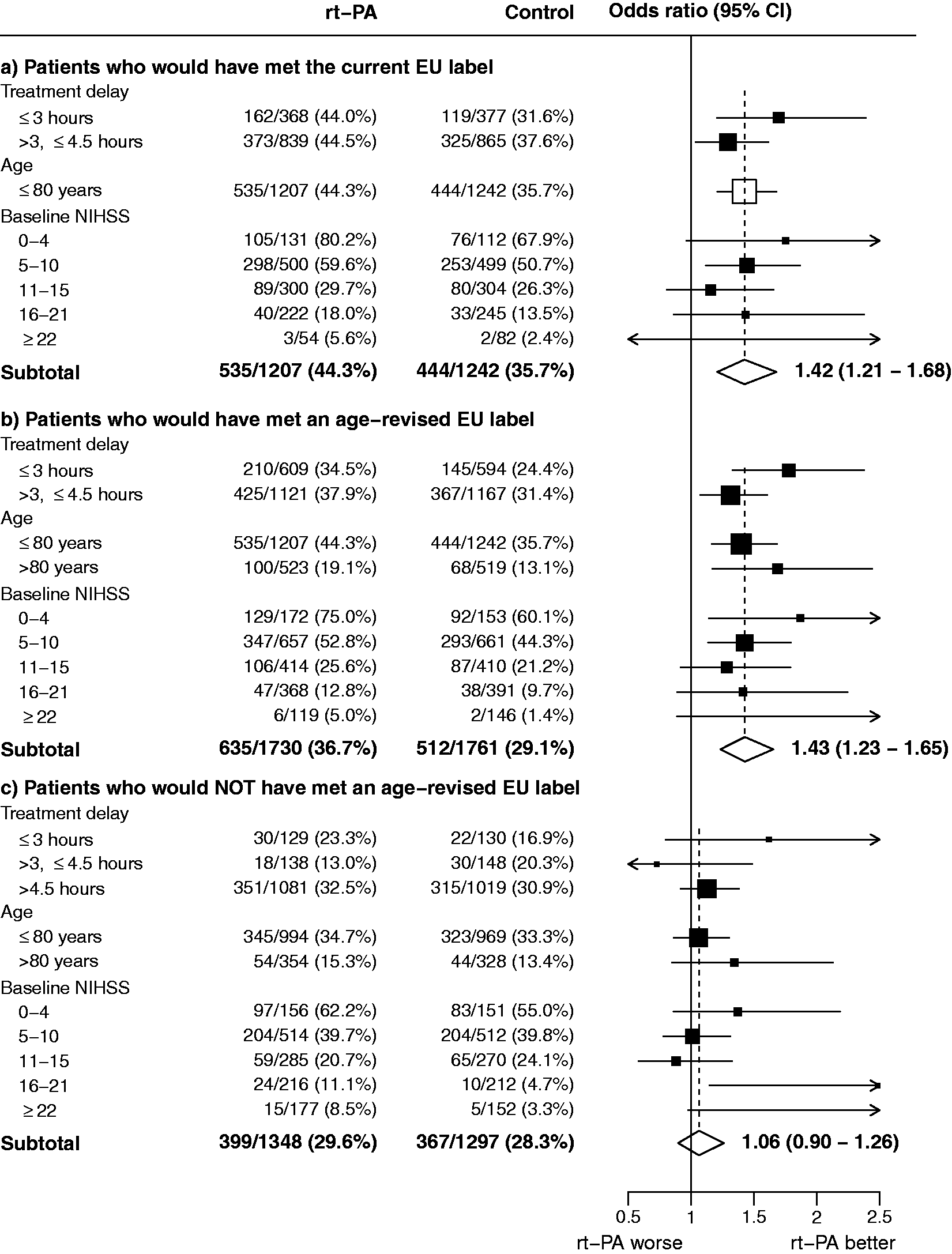
Effect of rt-PA on an excellent stroke outcome (mRS 0–1) in groups defined by the current European Union label as well as a European Union label without an upper age restriction. For (a–c), the odds ratios represented by open diamonds or open squares are derived from trial-stratified logistic regression estimates adjusted only for treatment allocation. By contrast, the odds ratios represented by filled squares are derived from trial-stratified logistic regression models which allow separate estimation of the OR at different levels of, respectively, treatment delay, age, and baseline NIHSS, with further adjustment for the other two baseline characteristics (but not for possible interactions with those characteristics). Consequently, the information-weighted average of the subgroup-specific estimates does not necessarily equal the summary odds ratios shown by the open diamonds (main effect estimates which are additionally adjusted for baseline treatment delay, age, and baseline NIHSS are shown in online Supplementary Table 4).
The odds for sICH were increased by alteplase: OR 5.25 (2.73–10.13) for parenchymal hemorrhage type 2; OR 5.87 (2.45–14.08) for the SITS-MOST definition; and OR 8.27 (2.47–27.64) for fatal ICH within seven days (online Supplementary Figures 2(a), 3(a), and 4(a)). The average absolute excess risk of fatal ICH within seven days was 1.7% (0.9%–2.5%) (online Supplementary Figure 4(a)). There was no evidence of an increase in 90-day mortality with alteplase: hazard ratio (HR) 0.98 (0.76–1.25; Figure 2).
Effect of rt-PA on 90-day mortality in groups defined by the current and extended labels in the EU and US. For both the EU and US label, and (a–c), Cox proportional hazards regression with stratification by trial and adjustment only for treatment allocation was used to estimate the hazard ratio and its 95% confidence interval for each period at risk. Patients can only contribute to a particular risk period if they have already survived any preceding periods. Denominators therefore reflect the numbers of patients at risk of death at the start of each shown period.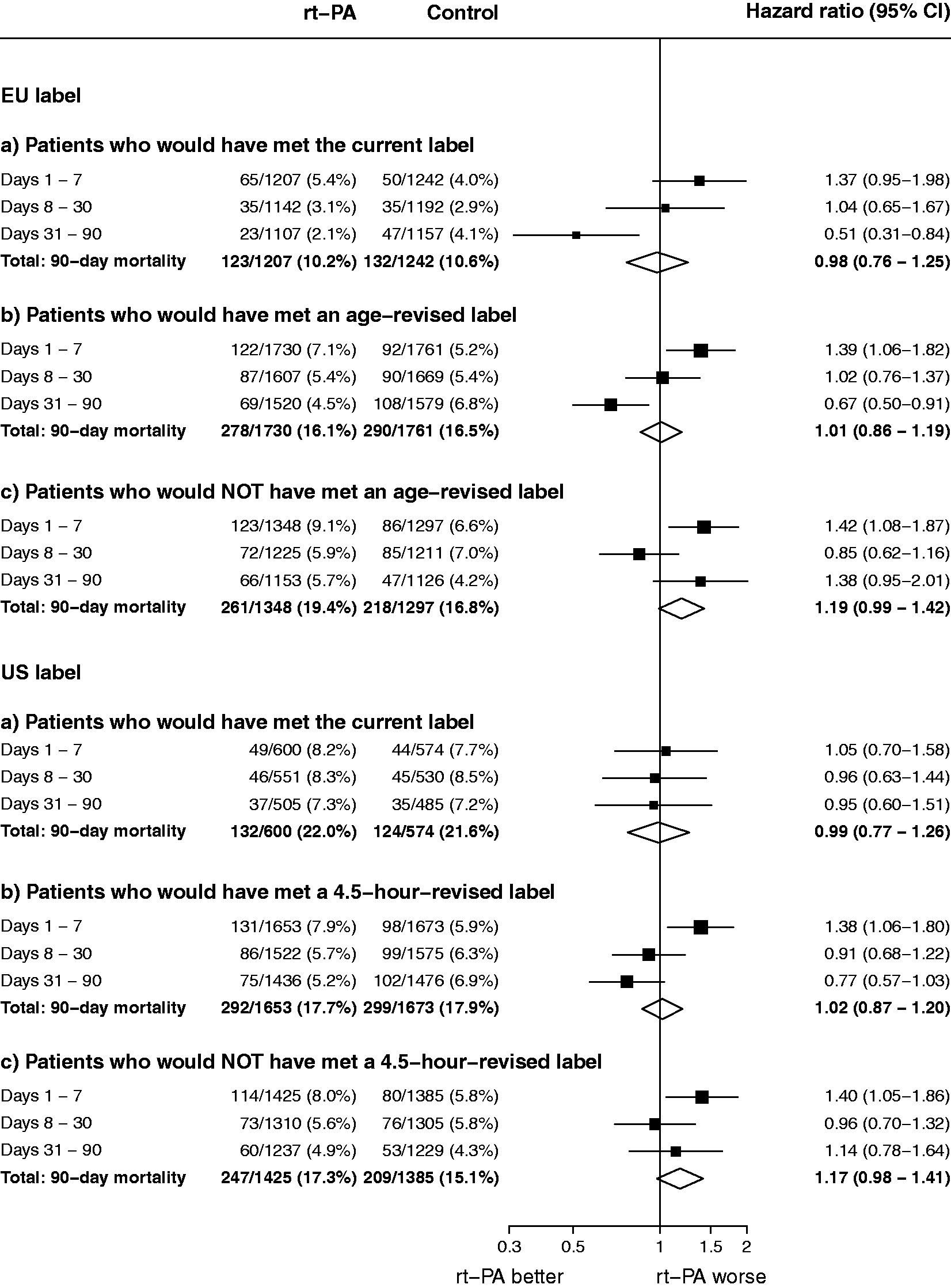
Effects among patients who would have met an age-revised EU label
Among 3491/6136 (57%) patients who would have met an EU label revised to include patients aged >80 (online Supplementary Tables 1 and 2), the odds ratios for achieving an excellent functional outcome and for gaining any improvement in mRS were 1.43 (1.23−1.65) and 1.29 (1.14−1.45), respectively (Figure 1(b) and online Supplementary Figure 1(b)). For excellent functional outcome, benefit was found independently in both the 0–3 h (OR 1.78, 1.33–2.38) and the 3–4.5 h (OR 1.31, 1.07–1.60) cohorts, and in those aged >80 years (OR 1.69, 1.17–2.44) (Figure 1(b)).
The odds of sICH were increased by alteplase: OR 5.23 (3.18–8.62) for parenchymal hemorrhage type 2, OR 6.87 (3.25–14.52) for the SITS-MOST definition and OR 8.75 (3.45–22.21) for fatal ICH within seven days (online Supplementary Figures 2(b), 3(b), and 4(b)). The average absolute excess risk of fatal ICH within seven days was 2.1% (1.3%–2.8%) (online Supplementary Figure 4(b)). There was no significant increase in 90-day mortality in this age-extended group (HR 1.01, 0.86–1.19; Figure 2). (Note that stratification by trial and the non-proportional influence of alteplase on survival generates this HR despite crude 90-day mortality being 16.1% with alteplase and 16.5% with control.)
Effects among patients who would not have met an age-revised EU label
Overall, among all patients who would not have met an age-revised EU label (online Supplementary Table 1), there was no significant effect on excellent outcome (OR 1.06, 0.90−1.26) nor for any mRS improvement (common OR 0.98, 0.85–1.12) (Figure 1(c) and online Supplementary Figure 1(c)). The odds of sICH were increased: for PH2 the OR was 7.37 (4.40–12.33), for SITS-MOST sICH 7.76 (3.69–16.29), and for fatal ICH within seven days 8.71 (3.44–22.03) (online Supplementary Figures 2(c), 3(c), and 4(c)). The average absolute excess risk of fatal ICH within seven days was 2.8% (1.9%–3.9%) (online Supplementary Figure 4(c)). There was an increase in early mortality (i.e. deaths within seven days) (HR 1.42, 1.08–1.87) which was not offset by improved longevity in survivors, giving an HR for 90-day mortality of 1.19 (0.99–1.42) (Figure 2).
Effects among patients who would have met the current US label
A total of just 1174/6136 (19%) patients from the eight trials would have met the current US label criteria because of the low number of patients treated within the 3-h time window (Tables 2 and 3). Among such participants, alteplase increased the odds of achieving mRS 0-1 (OR 1.55, 1.19–2.01) and of gaining any improvement in mRS (common OR 1.33, 1.09−1.63) (Figure 3(a) and online Supplementary Figure 5(a)).
Effect of rt-PA on an excellent stroke outcome (mRS 0–1) in groups defined by the current US label as well as a US label with a 4.5-h time window. For (a–c), the odds ratios represented by open diamonds or open squares are derived from trial-stratified logistic regression estimates adjusted only for treatment allocation. By contrast, the odds ratios represented by filled squares are derived from trial-stratified logistic regression models which allow separate estimation of the OR at different levels of, respectively, treatment delay, age, and baseline NIHSS, with further adjustment for the other two baseline characteristics (but not for possible interactions with those characteristics). Consequently, the information-weighted average of the subgroup-specific estimates does not necessarily equal the summary odds ratios shown by the open diamonds (main effect estimates which are additionally adjusted for baseline treatment delay, age and baseline NIHSS are shown in online Supplementary Table 4).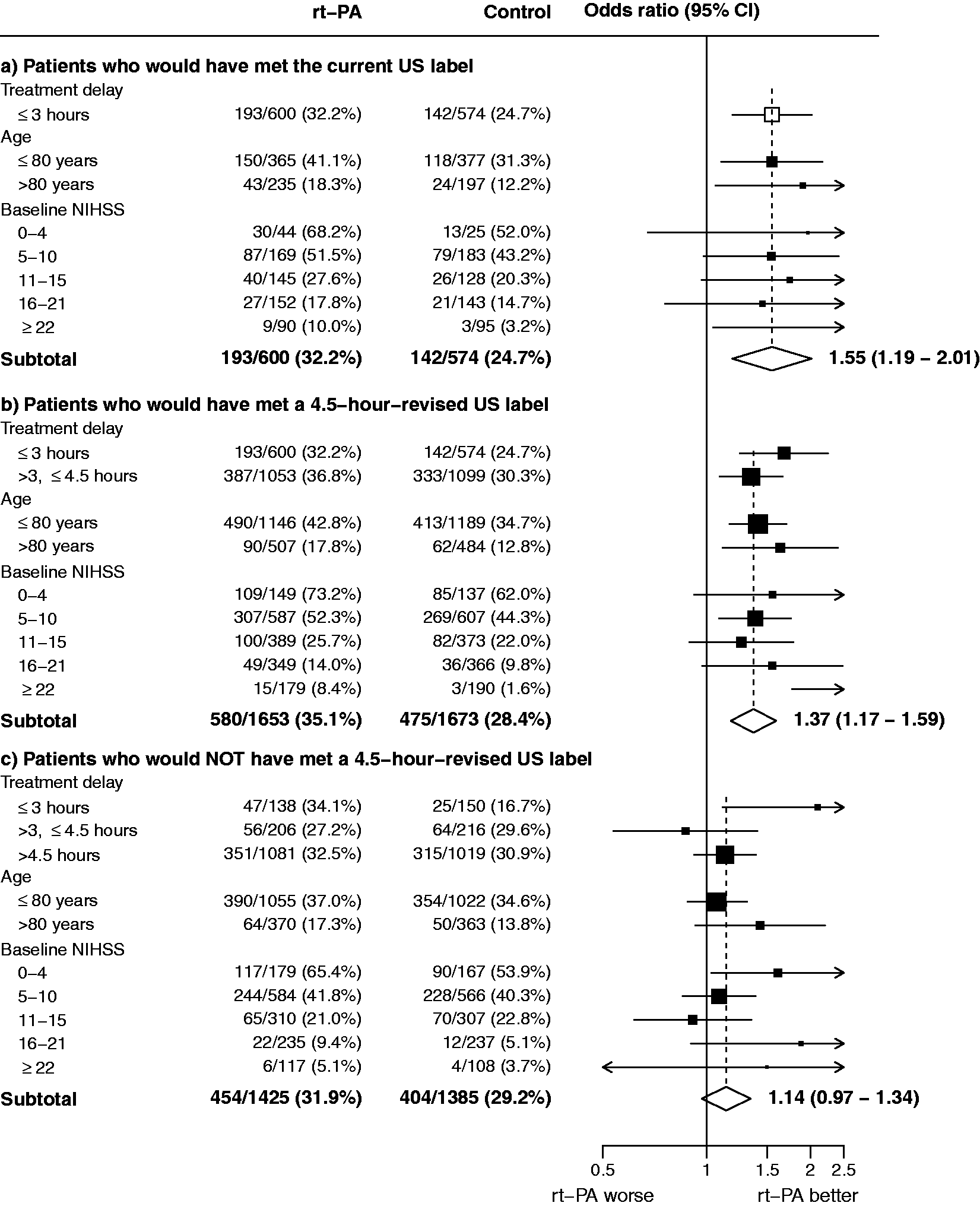
The odds of sICH were increased by alteplase: OR 6.38 (2.47–16.47) for PH2, OR 9.55 (2.22–41.06) for the SITS-MOST definition, and OR 15.19 (2.01–114.9) for fatal ICH (online Supplementary Figures 6(a), 7(a), and 8(a)). The average absolute excess risk of fatal ICH within seven days was 2.5% (1.2%–3.8%) (online Supplementary Figure 8(a)). Despite this increased early risk, there was no evidence of an increase in 90-day mortality with alteplase (HR 0.99, 0.77–1.26; Figure 2).
Effects among patients who would have met a 4.5-h-revised US label
Among 3326/6136 (54%) patients who would have met a 4.5-h-revised US label criteria, the odds ratios for achieving an excellent functional outcome and for gaining any improvement in mRS were 1.37 (1.17−1.59) and 1.24 (1.10−1.40), respectively (Figure 3(b) and online Supplementary Figure 5(b)). For excellent functional outcome, benefit was found independently in both the 0–3 h (OR 1.68, 1.25–2.25) and the 3–4.5 h (OR 1.35, 1.09–1.66) cohorts (Figure 1(b)).
The odds of sICH were increased by alteplase: OR 5.58 (3.35–9.30) for PH2, OR 7.35 (3.32–16.29) for the SITS-MOST definition, and OR 9.11 (3.60 – 23.07) for fatal ICH within seven days (online Supplementary Figures 6(b), 7(b), and 8(b)). The average absolute excess risk of fatal ICH within seven days was 2.3% (1.5%–3.1%) (online Supplementary Figure 8(b)). There was no significant increase in 90-day mortality in this cohort (HR 1.02, 0.87–1.20; (Figure 2).
Effects among patients who would not have met a 4.5-h-revised US label
Overall, among patients not satisfying a 4.5-h-revised US label, there was no evidence of benefit (OR 1.14 (0.97−1.34) for excellent functional outcome, common OR 1.03 (0.90–1.18) for any mRS improvement (Figure 3(c) and online Supplementary Figure 5(c)). The odds of sICH were increased: the OR was 6.89 (4.17–11.38) for PH2, 7.19 (3.56–14.50) for the SITS-MOST definition, and 8.32 (3.28–21.09) for fatal hemorrhage within seven days (online Supplementary Figures 6(c), 7(c), and 8(c)). The average absolute excess risk of fatal ICH within seven days was 2.6% (1.7%–3.5%) (online Supplementary Figure 8(c)). This early risk resulted in an increase in early mortality (i.e. days 1–7) (HR 1.40, 1.05–1.86) that was not offset by reduced mortality among those who survived the first week (HR for 90-day mortality 1.17 (0.98–1.41) (Figure 2).
Effects on the distribution of mRS scores
Figure 4 shows the distribution of the mRS scores at 3–6 months in each of the three cohorts (on label, on revised label, and off revised label) within
the EU and the US. Distribution of mRS at 3−6 months by randomized treatment allocation in groups defined by the current and extended labels in the EU and US. mRS score was ascertained at six months for IST-3 and three months for other trials.
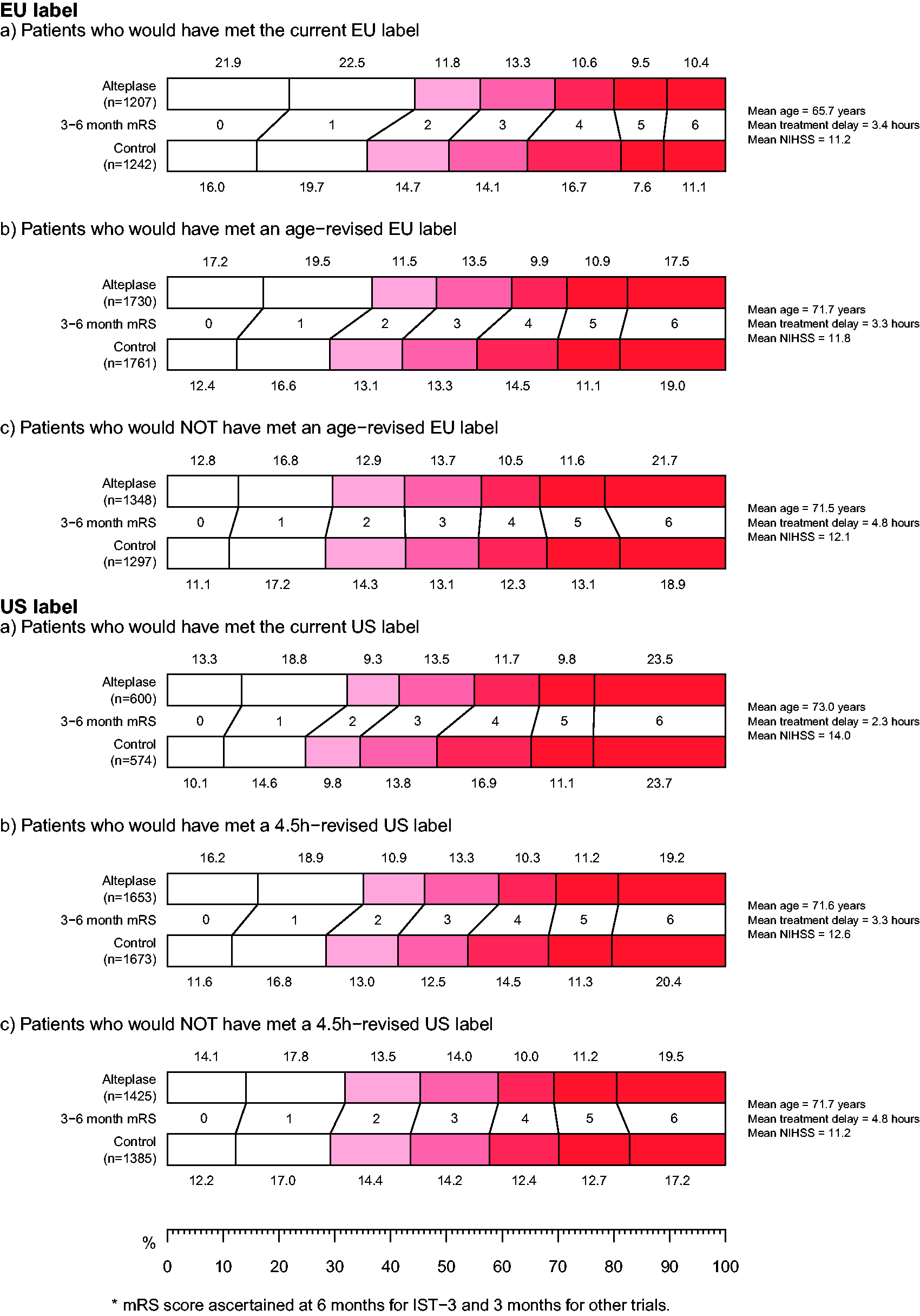
These distributions illustrate a similar pattern in the two regions: in both cases patient cohorts defined by the hypothetically extended labels derived clear net benefit, with a shift towards reduced disability and no significant excess of mortality.
Sensitivity analyses
For each of the contributing cohorts of patients, the baseline balance in key prognostic variables between those allocated alteplase and those allocated control is shown in online Supplementary Tables 3(a) to (f). As expected, randomization resulted in good balance for most characteristics in all cohorts. However, among the 1174 patients who would have met the current US label (online Supplementary Table 3(d)), there was a chance imbalance in mean age, with those allocated alteplase on average 1.8 years older than those allocated control. To assess the impact of this, treatment effect estimates for each cohort for each of the main outcomes were re-estimated after additional adjustment for baseline treatment delay, age and stroke severity (online Supplementary Table 4). For the cohort who would have met the US label, the adjusted OR estimates were 1.69 (1.26–2.28) for excellent functional outcome and 1.42 (1.15–1.76) for any upwards shift in mRS.
Discussion
The regulations that govern medicinal products seek to ensure that only products that meet standards of quality and safety, and have reasonable evidence of efficacy, are marketed. However, there are important differences between patient eligibility for intravenous alteplase treatment according to scientific guidelines and to marketing authorizations in both US and EU. The present analyses from the STT Collaboration indicate that patients who would have met the criteria of the AHA or ESO guidelines but would have been excluded by the regulatory authorization derived significant and clinically useful benefit by 90 days, with an early increased risk of death from intracerebral hemorrhage completely offset by a later improvement in stroke survival. Within the STT dataset, only two-fifths of patients (mean treatment delay 3.4 h) would have met the current criteria of the EU label, but this rose to over half (57%; mean delay 3.3 h) when the age criterion was removed (in accordance with ESO guidelines). For this extended group, alteplase yielded an excellent functional outcome with an OR of 1.43 (1.23−1.65), without any excess risk of 90-day mortality (16.1% with alteplase and 16.5% with control: HR 1.01, 0.86–1.19).
The current US label suggests a shorter time window for treatment than the EU label, and only 19% (mean delay 2.3 h) of the individuals in our dataset would have been included by it. A 4.5-h-revised version of the US label, however, would have been met by 54% of individuals (mean delay 3.3 h), and among these patients the effects of alteplase closely matched those of the age-revised EU label, with an odds ratio for achieving excellent outcome of 1.37 (1.17−1.59), again without any excess risk of 90-day mortality (17.7% with alteplase and 17.9% with control: HR 1.02, 0.87−1.20).
Among all patients treated with 0.9 mg/kg alteplase who would not have met revised labels in the EU and the US there was no significant functional benefit yet they suffered an overall hazard amounting to 2.2–2.6% excess mortality at day 90, due entirely to the excess in early fatal ICH. This is consistent with their long average treatment delay (4.8 h in both cases) together with the previously documented observation that the proportional benefits of intravenous alteplase (but not the risks) diminish with increased delay. These data indicate that patients failing to meet scientific guidelines for intravenous alteplase are not likely to benefit from alteplase after acute ischemic stroke.
The initial risk of intracerebral bleeding after intravenous alteplase is well recognized. 14 Despite this, there was, on average, an improvement in mRS among patients meeting the current or revised labels. Around half of the patients who suffered sICH survived beyond seven days. Even so, the distribution of functional outcomes by 90 days favors alteplase, implying that functional outcome, survival or both improved sufficiently in the remaining patients to offset this effect on the average outcome for the cohort (Figure 4). By 90 days, the excess risk of early death was offset by increased survival in the treated group. Although we should not disregard this early risk, it has been apparent from previous analyses that the absolute risk of serious intracerebral bleeding is higher among patients who present with severe, and thus likely disabling, stroke. The survival benefit persists among the treated group with longer periods of follow-up.12,15 Among patients who met either revised label, the revisions had limited impact on the risk of fatal intracerebral bleeding, since we found the relative and absolute risks were similar between the current and revised label cohorts and were well within the margin of error: absolute excess 2.1% (1.3%–2.8%) for the revised EU label (currently 1.7% (0.8%–2.5%)) and 2.3% (1.5%–3.1%) for the revised US label (currently 2.5% (1.2%–3.8%)).
Our analyses have limitations: the main one is that our quantitative estimates of treatment effect might not be replicable in an equivalent group of patients because the data from these trials to some extent have defined the labels and recommendations. Notwithstanding this caveat, the main observation that estimates of treatment effect were similar for patients eligible for current and extended labels remains valid. The validity of our analyses is enhanced by having access to all the relevant individual patient data, common definitions for most data items, and stratification of the analyses to retain any influence of individual trials on the outcome. Alignment of the US and EU labels on age and time window may not increase uptake of thrombolysis in routine practice by the proportions suggested from our dataset. The case mix of patients enrolled to the thrombolysis trials represents a combination of various eligibility criteria applied to a population case mix which itself will have changed over the 20 years since some of these trials were conducted. Based on a snapshot survey of 10,633 recent thrombolysis treatment registrations in the United Kingdom (UK) undertaken by one author (KRL, unpublished data), we estimate that 42% more patients would meet a 4.5-h-revised US label than the current US label, and 36% more patients would meet an age-revised EU label than the current EU label. These are likely to be conservative estimates, since some stroke physicians in UK may presently restrict their use of thrombolysis to the current EU label criteria. A recent analysis of 56,689 patients’ data from 597 sites registered to the SITS international registry over 6.5 years reported that if all patients were treated by using ESO guidelines, an additional 17,031 would receive alteplase, which translates into 1922 more patients with favorable three-month outcomes. 26
Summary
Hypothetical revisions of the treatment labels for alteplase after acute ischemic stroke, increasing the time window to 4.5 h for the US criteria and removing the upper age limit from the EU criteria, substantially increased the proportion of patients for whom treatment was of net benefit without elevating 90-day mortality. These revisions are in reasonable alignment with existing ESO recommendations on alteplase use. The available evidence indicates that the current US and EU marketing authorizations for the use of intravenous alteplase following acute ischemic stroke are unduly restrictive and may well be contributing to unnecessary disability.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KRL reports fees and expenses from American Stroke Association, Applied Clinical Intelligence, Atrium, Boehringer Ingelheim, EVER NeuroPharma, Hilicon, Nestlé, Novartis, Servier, and Stroke Academic Industry Roundtable, and research funding to the University of Glasgow and to the Virtual International Stroke Trials Archive from Genentech, and is past president of the European Stroke Organisation. CB and JE have not accepted fees, honoraria, or paid consultancies but are, or have been, involved in clinical trials funded by Merck, Novartis, Pfizer, the Medicines Company, and Boehringer Ingelheim. GA has received fees for consultancy from Medtronic and iSchemaView, and owns stock in iSchemaView. EB is employed by Boehringer Ingelheim. SMD has received honoraria from AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Medtronic, and Pfizer. GAD is co-principal investigator for the EXTEND trial using alteplase and has received honoraria from Boehringer Ingelheim, Bayer, Pfizer, Sanofi, and Merk Sharp & Dohme. JCG has acted as a consultant for Frazer Ltd and Stryker, has received grant support from the American Heart Association, Genentech, and Behring and has received grant support within the past three years from Haemonetics and Medtronic. MKa reports fees and expenses from H. Lundbeck A/S, Mitsubishi Pharma Europe, Siemens AG. RvK reports fees from H. Lundbeck A/S, Boehringer Ingelheim, Covidien, Brainsgate, Synarc, and Penumbra, Inc. RIL has received honoraria from Boehringer Ingelheim, Covidien, and Pfizer. JMO has received honoraria from Astra Zeneca, Boehringer Ingelheim, Bristol Myers Squibb, Pfizer, and Servier. MP has received travel support from Boehringer Ingelheim. PAGS declares that he was the Chief Investigator of the IST-3 trial, funded by the Medical Research Council, the Stroke Association and the Health Foundation, the IST-3 pilot study was supported by a donation of drug and placebo from Boehringer Ingelheim, and PAGS has received Honoraria and Travel expenses paid to his Department from Boehringer Ingelheim. DT reports honoraria from Boehringer Ingelheim, Bayer, and Pfizer. KT has received research grant support from the Japan Agency for Medical Research and Development and fees from Mitsubishi Tanabe Pharma. JMW declares trial funding from the Medical Research Council, Efficacy and Mechanism Evaluation Programme, Stroke Association, and Health Foundation. NW's institution has received research grants from Boehringer Ingelheim, Covidien, Stryker, and Codman. WNW was funded by a Medical Research Council Clinician Scientist Fellowship (G0902303). WH reports honoraria from Boehringer Ingelheim, Daiichi Sankyo, and Bayer, receipt of an unrestricted research grant from Boehringer Ingelheim to perform the ECASS-4 EXTEND trial, and past chairmanship of the ECASS 1–3 thrombolysis trials. WH is also current president of the World Stroke Organisation. LB, TB, GC, GdZ, GH, MKo, MGL and PL declare no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Stroke Thrombolysis Trialists’ Collaboration is coordinated by the MRC Population Health Research Unit, which is part of the Clinical Trial Service Unit & Epidemiological Studies Unit at the University of Oxford, UK. The Unit receives core funding from the UK Medical Research Council and the British Heart Foundation. This work also received support from the University of Glasgow and University of Edinburgh.
Appendix 1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.