
Abstract
During the COVID-19 pandemic, the race to find an effective vaccine or treatment saw an ‘extraordinary number’ of clinical trials being conducted. While there were some key success stories, not all trials produced results that informed patient care. There was a significant amount of waste in clinical research during the pandemic which is said to have hampered an evidence-based response. Conducting trials which could have been predicted to fail to answer the research question (e.g. because they are not large enough to provide a definitive result) is not only a waste of resources but also a breach of research participants’ trust and a violation of research ethics.
The issues seen in COVID-19 clinical trials are symptomatic of a wider trial design crisis where many trials do not provide informative results. This paper examines the roles of key stakeholders in delivering ethical and informative trials and whether guidance published by ‘The Good Clinical Trials Collaborative’ could be used to align key stakeholder groups and enable a joined-up approach to improve clinical trial design.
Introduction
Large, randomised clinical trials were crucial during the COVID-19 pandemic to provide reliable evidence about the potential benefits and harms (or lack thereof) of interventions for COVID-19 (Pessoa-Amorim et al., 2021). The race to find a vaccine or effective treatment saw an ‘extraordinary number’ of trials being conducted (Glasziou et al., 2020), supported by fast track procedures from ethics committees and regulators (Health Research Authority, 2022).
While there were important success stories, such as the accelerated development of vaccines (Bok et al., 2021), and trials such as RECOVERY and SOLIDARITY being set up in record time (Tikkinen et al., 2020), not all trials produced results that informed patient care. There was a significant amount of waste in clinical research due to poorly designed, uninformative and duplicative trials, which is said to have hampered an evidence-based response (Glasziou et al., 2020). An analysis of clinical trials for COVID-19 worldwide performed by United States Food and Drug Administration (FDA) scientists found that of 2895 trial arms examined, approximately 5% could be considered randomized and adequately powered, and only about a quarter of enrolled patients contributed to adequately powered and well-controlled trials (Bugin and Woodcock, 2021). Conducting trials which could have been predicted to fail is not only a waste of resources, but also a breach of research participants’ trust and a violation of research ethics (Zarin et al., 2019), particularly given the backdrop of scarce healthcare resources, social distancing measures and lockdowns during the pandemic.
To solve this problem, it is important to define what makes a trial uninformative and how we can recognise an informative trial: ‘An uninformative clinical trial is one that fails to produce meaningful results for patients, clinicians, researchers, or policy makers’ (Morrell et al., 2023: 99). For a trial to be considered informative, London and Kimmelman suggest that research should embody five conditions of informativeness and social value (London and Kimmelman, 2020). These are:
Importance – trials should address key evidence gaps and a public health concern.
Rigorous design – trials should detect meaningful effects.
Analytical integrity – analyses should be prespecified and publicly available.
Reporting - trials should be reported in full, promptly, and consistently with prespecified analyses.
Feasibility – trials should be feasible in terms of both the recruitment targets and the operational aspects of the protocol.
The importance of high quality clinical research was recognised during the pandemic, with the UK government (Department of Health and Social Care, 2021; G7, 2021; O'Shaughnessy, 2023), European Medicines Agency (EMA) (European Medicines Agency, 2022) and the World Health Organization (WHO; Moorthy et al., 2024) launching policy initiatives to improve the clinical trial landscape.
To ensure ethical informative trials, key stakeholders, including Research Ethics Committees (RECs), funders, regulators and sponsors must adopt a common approach. Guidance published by The Good Clinical Trials Collaborative (GCTC) aims to address this challenge by providing principles-based standards for the design, conduct and reporting of clinical trials that are relevant and accessible to all stakeholders (GCTC, 2022).
This paper takes a UK perspective in examining the role of key stakeholders for the identification of uninformative trials as well as areas for improvement. Nevertheless, the findings have global relevance.
Clinical trials – a design crisis?
Whilst the extent of uninformative research was highlighted during the COVID-19 pandemic, it is not a new problem. A recent study found that just 26% of heart disease, diabetes and lung cancer trials in the US informed clinical practice. Consequently, more than 30% of participants were enrolled in uninformative trials (Hutchinson et al., 2022). Back in 2009, Chalmers and Glasziou reported that up to 85% of overall research investment is wasted (Chalmers and Glasziou, 2009).
Rigorous design is a key condition of informativeness and avoiding research waste (Chalmers and Glasziou, 2009; London and Kimmelman, 2020). However, some researchers believe that the rules and regulations governing trials have become a barrier to conducting well-designed, informative trials (Califf, 2006; Collins et al., 2020; Bowman et al., 2022). Whilst intended to improve participant safety and the reliability of results, some have argued that they instead draw attention away from the underlying scientific principles of randomised trials, towards disproportionate and rigid interpretation of available guidelines (Collins et al., 2020; Janiaud et al., 2021), that has in turn led to an increase in the cost and complexity of conducting clinical trials.
Adequately sized, randomised controlled trials are broadly considered to be the gold-standard for generating reliable evidence and essential wherever there is uncertainty over the relative benefits or harms of a treatment (Collins and MacMahon, 2001; Collins et al., 2020; Bowman et al., 2022). To inform patient care reliably, trials must be large enough (i.e. have sufficient statistical power) to differentiate between a treatment with modest effects (beneficial or harmful) and one with no meaningful effect at all. For example, the RECOVERY trial of treatments for COVID-19 found that low-dose dexamethasone reduced 28-day mortality for those on mechanical ventilation by about one-third (Horby et al., 2021). This evidence was sufficiently clear to be implemented in the UK within hours of becoming available, and implemented globally within weeks (Pessoa-Amorim et al., 2021); it is estimated to have saved over a million lives worldwide (Stokel-Walker, 2021). Beyond COVID-19, there are many examples of well-designed trials for a range of health issues (such as heart disease, stroke and breast cancer) demonstrating moderate treatment benefits that have led to millions of lives being saved or improved (Neal et al., 2000).
Despite this, there has been a trend towards the use of smaller trials and non-randomised observational studies in recent years (Eisenstein et al., 2008; Collins et al., 2020; Bowman et al., 2022). As noted by Califf et al. (2012), small trials are appropriate in many cases, such as earlier phase drug evaluations and investigations of biological or behavioural mechanisms (Califf et al., 2012). However, the inappropriate use of small trials has led to a reliance on surrogate and composite endpoints rather than on simple, clinical endpoints (Janiaud et al., 2020). Restrictive eligibility criteria are commonplace and can increase complexity, deter participation, and reduce the reliability of their results (Janiaud et al., 2021). In addition, many trials limit the duration of follow-up which can lead to an incomplete understanding of the long-term effects of the intervention (Bowman et al., 2022). As a result of these design choices, trial results may not be sufficiently clear to guide clinical practice and patient care, highlighting the ethical implications of poor design.
The quality of trial design can have a significant impact upon recruitment; if the trial is overly complex and not accessible to potential participants, even (planned-for) large-scale research is likely to be underpowered due to the inability to recruit or retain participants. Adequately sized randomised trials can be inappropriately designed if they fail to consider the context of the trial and operational feasibility (Mhlongo, 2024). It is vital that the trial design minimises burdens on participants and considers the environment in which the intervention is intended to be used to ensure the trial is fit for purpose.
Well-designed trials are essential, particularly in a crisis, to minimise the burden on overstretched healthcare professionals (Pessoa-Amorim et al., 2021), but they are also essential to ethical trial participation. Many researchers and ethicists argue that it is unethical to enrol participants into a study that has little chance of reaching conclusive results (Califf et al., 2022). For instance: Science and ethics do not conflict; valid science is an ethical requirement. Unless research generates reliable and valid data that can be interpreted and used by the specified beneficiaries of the research, it will have no social value, and participants will be exposed to risks for no benefits (Emanuel et al., 2004: 933).
There is a clear need to increase focus on conducting well-designed clinical trials that can produce informative results to improve public health outcomes and reduce waste in clinical research.
Uninformative research – reviewer roles
Clinical trials are highly regulated but compliance with regulatory requirements does not ensure that the trial will produce informative results. In their report from a qualitative interview study, Morrell et al. (2023) highlighted concerns around a lack of a systematic approach to determining scientific value and feasibility, including who should be responsible for the review and exactly when it should be conducted (Morrell et al., 2023).
In this section we examine the current approach to scientific and ethical review for clinical trials in the UK prior to the start of a trial where there is a potential to identify and prevent uninformative trials. The key stakeholder groups involved in each type of review include funders, peer reviewers, sponsors, regulators, and research ethics committees (RECs).
Funders
Clinical research can be funded through commercial or non-commercial routes. Many non-commercial funders require open competition for grant funding. However, funders have faced criticism over the way in which research quality has been assessed with more focus being placed on meeting metrics and milestones than on producing informative results (McLean et al., 2018). Similarly, the Wellcome Trust reports that a focus on publication in the competitive funding process has contributed to a culture of ‘publish or perish’ (Wellcome Trust, 2020). The pressure to publish has also been identified as a potential driver for a decrease in the quality of research as well as a potential driver for research misconduct (Paruzel-Czachura et al., 2021).
Research ethics committees
The purpose of research ethics review is to protect the rights, safety, dignity and wellbeing of research participants (Health Research Authority, 2020). A key aspect of research ethics review is to weigh the perceived benefits and risks for research participants and society against the value of the knowledge expected to be produced. Therefore, as asserted by Emanuel et al. (2004), research that does not produce reliable and useable data brings burdens to participants without benefits.
Despite the clear ethical implications of poorly designed research, the attempts by many RECs to seek assurances on the designs of research projects has been labelled as ‘ethics creep’ (unwarranted interference and beyond remit; Haggerty, 2004; Gunsalus et al., 2006). This forms part of a wider narrative of dissatisfaction with REC review, with researchers raising concerns around lack of proportionality (Tsampiras and Müller, 2018), unnecessary bureaucracy (Loveday and Mitchell, 2010) and lack of consistency (Randerson, 2006). It has also been argued that the REC review process itself is difficult for researchers to engage with, for example, due to a perceived lack of transparency and misunderstandings around the requirements that need to be met (Johnsson et al., 2014).
This issue is further compounded by questions over whose responsibility it is to ensure the scientific validity of clinical research. RECs in the UK are not expected to verify that the design of a clinical trial is appropriate. The Health Research Authority (HRA), which is responsible for RECs associated with NHS Research, states in their ‘Governance arrangements for research ethics committees’ that: A REC need not reconsider the quality of the science, as this is the responsibility of the sponsor and will have been subject to review by one or more experts in the field (known as ‘peer review’). The REC will be satisfied with credible assurances that the research has an identified sponsor and that it takes account of appropriate scientific peer review (Health Research Authority, 2020: 26).
In contrast to the approach in the UK, Binik and Hey (2019) assert that RECs have a fundamental role in assessing the scientific value of clinical trials and propose a framework for RECs to assess both the scientific value and validity of the research.
Binik and Hey (2019) argue that the REC itself needs to have sufficient expertise to scrutinise a study’s design to evaluate the associated benefits and risks properly. This requires understanding of whether the proposed trial is likely to answer the research question and also assessing whether the risks to participants have been minimised throughout its design. However, it is important to recognise that RECs may not be able to ensure a sufficient level of scientific expertise to conduct a proper assessment of scientific validity and feasibility, particularly when novel and unconventional trial designs are utilised.
Gelinas et al. (2023) conducted a Delphi working group on how to limit uninformative trials which concluded that Institutional Review Boards (IRBs; and therefore, also RECs) have a responsibility to ensure scientific value. They conclude that the IRB’s assessment of the scientific value should be linked to a landscape analysis conducted by the sponsor or principal investigator, which can be used to assess whether the risks to participants are reasonable in light of the potential knowledge to be gained. The working group also highlighted the need for overarching standards for scientific review, and overall conditions for clinical trials to be deemed as scientifically and ethically justifiable. Their findings emphasise the need for a multi-stakeholder approach, especially in relation to the ongoing assessment of scientific validity.
Sponsors
The sponsor of a clinical trial is the person, institution or organisation who takes overall responsibility for the design and conduct of the research, including ensuring compliance with relevant regulatory requirements (Health Research Authority, 2017; UK.SI.1031, 2004). In the UK, the sponsor is expected to ensure the quality of research prior to submission for ethics and regulatory approval. The UK Policy Framework for Health and Social Care Research outlines the requirements for research sponsors, which includes identifying and addressing poorly designed or planned research, ensuring that prior research is taken into account, that there is appropriate use of patient and public involvement, and that studies are scientifically sound, safe, ethical, legal, and feasible.
However, it is important to acknowledge that a sponsor will have an active interest in the research taking place and instances of ‘sponsorship bias’ are well documented (Jefferson, 2020). In addition, there are widely different processes and approaches to sponsorship between commercial and non-commercial sponsors (Ravinetto et al., 2015). Without an overarching framework or guidance, any required assessments by sponsors are likely to vary significantly.
Peer reviewers
In the UK, for any research taking place in an NHS or social care setting, the Health Research Authority requires the sponsor to ensure the research is scientifically sound through independent expert review (Health Research Authority, 2017). Peer review is also utilised in competitive funding processes internationally to assess the scientific quality of proposals. However, the peer review process has been repeatedly called into question, with issues reported around the independence, subjectivity and accuracy of reviews (Smith, 1988; Rowland, 2002; Davidoff, 2004; Smith, 2006; Henderson, 2010; Science and Technology Select Committee, 2011).
Regulators
Regulators, such as the European Medicines Agency and the UK Medicines and Healthcare Regulatory Authority, also have a key role in reviewing and approving clinical trials. Their current approach focuses on the requirements for clinical trial authorisation in accordance with the relevant regulations in that country.
However, clinical trial authorisations are focused solely on scientific criteria to determine whether the medicines being investigated meet the necessary quality, safety and efficacy legislative requirements (European Medicines Agency, 2023). This review does not incorporate consideration of operational feasibility or the ability to recruit the required number of participants.
In summary, there is a gap in the current approach to reviewing clinical trials. Assessment of their informativeness falls beyond the scope of the responsibilities of key stakeholders.
Improving the process
Whilst there is a clear need for an overarching set of standards and a multi-stakeholder approach to the prevention of uninformative clinical trials, the responsibilities of each stakeholder group in this regard are not well defined. All stakeholder groups could be said to have a collective responsibility to ensure that trials are ethical and informative, but without addressing gaps within and between these governance processes, poorly designed trials will continue. An agreed and shared framework, as well as clearly defined responsibilities for each group might help to identify stages in a trial’s lifecycle where the risks of uninformativeness can be identified and addressed.
Nevertheless, it is important to recognise that a balance is required between preventing uninformative trials and adding layers of bureaucracy or duplication that might delay or prevent worthwhile research from taking place. At the 75th World Health Assembly, the WHO called for action to be taken to strengthen clinical trials to ensure high-quality, ethical research to achieve international health-related development goals (World Health Organization, 2022). The resolution called on member states to improve their infrastructure and ability to prioritise well-designed informative trials, and recommended further support for RECs and regulatory authorities to implement efficient governance processes that support well designed and well implemented trials (World Health Organization, 2022).
Recognising the need to encourage robust assessment of clinical trials whilst enabling flexibility and innovation, the WHO developed draft guidance for public consultation on best practices for clinical trials (World Health Organization, 2023). The guidance focuses on three key areas: key scientific and ethical considerations for good, controlled trials (Section A), guidance on strengthening the clinical trials ecosystem (Section B), and on addressing under-represented populations (Section C). Section A of the guidance is based on the Good Clinical Trials Collaborative guidance for Good Randomized Clinical Trials.
The Good Clinical Trials Collaborative is a global initiative which co-developed new, proportionate and flexible guidance for the conduct of randomised clinical trials with a broad range of international stakeholders across multiple resource settings (high, middle and low income countries; The Good Clinical Trials Collaborative, 2022). London and Kimmelman’s (2020) five principles of informativeness and social value are reflected and reinforced in the GCTC Guidance, which discusses how to operationalise these principles in greater detail, focusing on the issues that materially matter to the wellbeing of trial participants and the reliability of results.
The guidance is based around five principles, as shown in Figure 1.
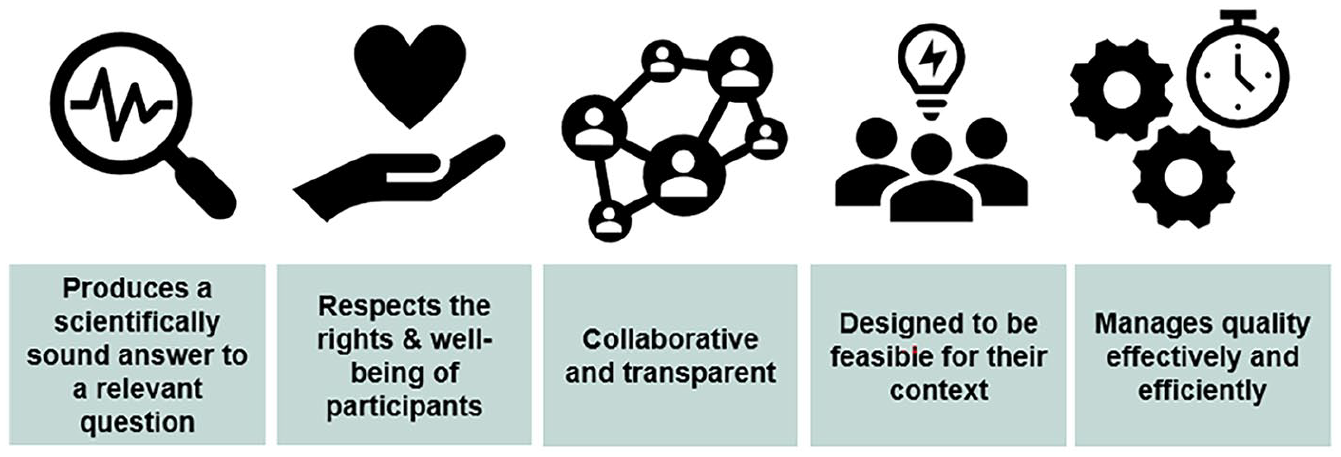
Good clinical trials collaborative principles.
The first WHO clinical trials forum, focusing on the future of the global trial ecosystems, identified that delivery of broad-based training for all stakeholders is required to support the effective implementation of agreed guidance on improving trial design (Moorthy et al., 2024).
However, as identified by Morrell et al. (2023), there is often disagreement amongst the research community about who the most appropriate stakeholders are to ensure scientific validity and feasibility. For example, not all participants agreed that the IRB should be involved, and some identified a lack of shared standards as being the main cause of shortcomings in existing processes for scientific review. Scientific review was considered to be a shared responsibility, with primary responsibility being held by the principal investigator and secondary responsibility being held by the various stakeholders responsible for reviewing the research.
The GCTC guidance is well placed to act as the agreed and shared framework for well-designed clinical trials across all stakeholder groups. As well as being incorporated into the WHO’s guidance on best practices for clinical trials (World Health Organization, 2023), it has been recognised by leaders in the cardiovascular field (Bowman et al., 2022). The GCTC guidance takes a principles-based approach and is intended to be applicable to any randomised trial, with any intervention, in any setting, and to enable use by a diverse range of stakeholders involved in the planning, conduct, analysis, interpretation, funding, and oversight of randomised clinical trials. However, the specific responsibilities of each stakeholder group to ensure well-designed, ethical trials need to be appropriately interpreted and implemented to fit the context of the trial in question. This has the advantage of the guidance being universally relevant but reinforces the conclusion from the first WHO clinical trials forum that effective training is crucial for successful implementation.
The responsibilities of each stakeholder group were explored by Gelinas et al. (2023) in their Delphi working group, which achieved consensus on conditions for ‘informativeness’ (which are aligned with the GCTC guidance) and agreed recommendations relating to; standards for scientific review, the role of the IRB, the role of the data monitoring committee and responsibilities during trial conduct. For example, they recommended that the sponsor and the principal investigator are responsible for performing a landscape analysis which is reviewed by the IRB. The results of this study also provide a useful insight into how the REC can perform its role of weighing risks and benefits of the study by using initial assessments performed by the principal investigator or sponsor.
Another important factor to be considered at the design stage is how emerging information about benefits and harms can be monitored from the trial data and other sources, including publications of relevant studies. As recognised by Bierer et al. (2023), the COVID-19 pandemic highlighted existing issues around how trials monitor a shifting evidence base. New evidence can render well-designed trials uninformative and there should be effective mechanisms in place to review the external evidence periodically. The GCTC guidance includes guidance on the importance of planning for monitoring of, and responding to, emerging information from appropriate sources within and outside the trial. Principle 1 of the guidance outlines high-level responsibilities of trial personnel and the data monitoring committee in this area.
To improve the clinical trials landscape, informative trials should be developed in accordance with a shared and agreed framework with clear responsibilities that are understood by all involved in the design, review and conduct of clinical trials. Any attempts to improve the quality of clinical trials should focus on ensuring that they are designed to be informative from the outset, rather than relying on regulators or ethics committees to identify flaws in design. This approach would not only increase transparency, but also reduce previously encountered frustrations with REC approvals and clinical trial governance systems.
In the examples outlined by Gelinas et al. (2023), the responsibility for identifying and evidencing the importance and relevance of the research question remained with the investigator and sponsor, but with full transparency for the IRB to assess the ethical implications of the research. This approach recognises the strengths of each stakeholder group without implementing a process of unnecessary overlapping or duplicative reviews. Adoption of the GCTC guidance could facilitate well-designed trials from the outset, with clear priorities for principal investigators, sponsors and peer reviewers to direct efforts to ensure the scientific validity and feasibility of the trial. Application of the guidance across these stakeholder groups and transparency of the reviews previously undertaken, would allow ethics committees and regulators to conduct their reviews effectively with additional assurances that any previous reviews had been conducted in accordance with an agreed framework. This would represent a significant improvement on the current approach in the UK where the REC is asked to take assurances from sponsors and peer reviewers without a clear insight into or an understanding of the criteria used to assess scientific validity or appropriateness of design.
An overview of how the guidance might be used by each stakeholder group is outlined in Table 1.
Stakeholder use of the GCTC guidance.

Conclusion
There is a clear need for well-designed, informative clinical trials to improve public health in pandemic settings and more broadly. However, the adverse ethical implications of poorly designed or uninformative clinical trials should not be understated.
As recognised by WHO, the UK Government and the European Medicines Agency, a coordinated approach is required to improve the clinical trials landscape. This includes adoption of a shared framework with clear responsibilities for all stakeholders to facilitate well-designed, informative trials.
We recommend that the GCTC Guidance is adopted by all stakeholders involved in the design, review and conduct of trials to ensure consistency and transparency regarding the requirements for informative trials. This principles-based guidance can be applied flexibly and consistently. If implemented as a shared framework across stakeholder groups it could facilitate identification of shared responsibilities (overlaps) and potential gaps in accountability to help ensure that the key requisites for informativeness are addressed.
Suggestions for implementation of this approach have been outlined (Table 1), emphasising that the overall responsibility for trial design remains with the sponsor and the principal investigator. Nevertheless, a shared framework and responsibilities for assessing a trial’s scientific validity and feasibility could help to promote both transparency and efficiency. This might, in turn, help to address recommendations from the World Health Organization to reduce the number of uninformative trials, to ensure better use of resources and avoid the ethical violations associated with uninformative trials.
Footnotes
Acknowledgements
We would like to thank Nick Medhurst, Lead for the Good Clinical Trials Collaborative for reviewing this article.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Emma Law and Isabel Smith are employees of Protas, a not-for-profit organisation focused on improving the quality of clinical trials. Protas has received funding from Sanofi, Regeneron, Moderna, NHS England, Schmidt Futures, Google Ventures, Flu Lab, Wellcome, and the Bill & Melinda Gates Foundation.
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.
Ethical approval
None required.