
Abstract
This article describes our efforts to screen and enrol clinical trial participants conscientiously in the COVID-19 pandemic setting. We present the standard screening and enrolment process prior to, and our process of adapting to, the pandemic. Our goal was to develop a way to screen and enrol people for clinical trials that was both equitable and effective. In addition, we outline the steps our research department took to ensure that ethical, clinical and logistical factors were considered when matching a patient to a clinical trial.
Background
The challenge of balancing research with a clinician’s duty of care is not a new one. Research ethics boards are responsible for ensuring the protection of research participants by assessing the risks versus the potential benefits (Weijer, 2000). However, evaluating a new disease added complexity since recommended treatments changed frequently, limited information was known about the disease, and new treatments were being tested.
The COVID-19 pandemic brought unprecedented challenges for researchers regarding clinical trial screening and enrolment. Finding solutions was complicated since the disease spread rapidly worldwide with no known treatments. Although ethical concerns about handling new diseases, resource allocation, misinformation, mistrust and changes in best practices existed before the pandemic, they became more pronounced in the context of the pandemic (see Table 1). Research staff had to assess each COVID-19-positive patient critically and practically to determine the best course of action for study enrolment.
New and exacerbated considerations during the COVID-19 pandemic.

The United States government began a concerted effort to fund and fast-track clinical trials to combat COVID-19. This effort meant that COVID-19 clinical trials proceeded with all the usual guidelines for new investigational products but within a compressed timeline. What usually took years or decades was accomplished in weeks or months. Because of this, research coordinators had to adapt their screening and enrolment procedures. Before the pandemic, studies might have had days or weeks to screen and enrol patients. The acute nature of COVID-19 and timing constraints necessitated immediate enrolment within hours of diagnosis.
In a clinical trial, the standard of care (SOC) is a reference point against which the new medications being tested can be evaluated for efficacy and safety. The World Medical Association (2013) has emphasised the necessity of a minimum SOC to guarantee clinical trial safety and ethical conduct. This standard helps to protect participants by providing a baseline for comparison of the effectiveness and safety of the new treatment compared to existing treatments or placebo. This ensures that the trial’s control group will receive the same quality of care and outcomes as they would outside the trial (Marouf and Esplin, 2015). The SOC and recommended treatments for the COVID-19 pandemic changed frequently during the pandemic. Treatment guidelines and protocols had to be frequently updated to incorporate new knowledge as more information became available about the virus’s mechanism of action and sequelae. Researchers and providers were forced to adjust and rule out specific treatments as new evidence emerged, which presented a significant challenge.
Repurposed medications for other diseases were tried as the SOC. Healthcare providers relied on preliminary data from China and European nations for treatment options, although most of these studies were observational rather than controlled clinical trials (Dos Santos, 2020). Healthcare providers selected treatments they knew to be effective (at least in vitro) against earlier viruses such as SARS and MERS but were not approved for COVID-19. Hydroxychloroquine, an anti-malaria medication, and lopinavir-ritonavir, an HIV treatment, were among the drugs that were evaluated but ultimately determined to be ineffective (Lam et al., 2020). Patients receiving information from multiple sources (social media, news sites, anecdotal, etc.) also tried self-repurposing of medications that were unapproved for COVID-19, such as ivermectin, an anti-parasitic medication (US Food and Drug Administration, 2021). As these and other drugs were being tried off-label by providers at the bedside and by patients at home, researchers were actively excluding these individuals from participating in clinical studies due to the potential confounding effects.
Equipoise refers to genuine uncertainty or reasonable doubt about the comparative effectiveness of different treatment options. Researchers must maintain equipoise in clinical trials when designing and conducting studies to ensure unbiased results. All treatment options being compared in a trial should be considered equally valid, and there should not be any predetermined preference for one treatment over another. By maintaining equipoise, researchers can avoid biases and ethical concerns when evaluating new treatments’ efficacy and safety (Cook and Sheets, 2011). Equipoise was difficult when the SOC was absent, changed, or based upon less rigorous data.
Screening
Screening a potential study participant is essential for ensuring participant safety, answering important clinical questions and enhancing study validity and generalisability. Eligibility criteria refer to the set of conditions or requirements that must be met to be eligible to participate in a clinical trial. Having clinical trials competing to enrol from the same patient populations can lower the likelihood of meeting enrolment targets, which causes the study to be underpowered and reduces the ability of the study to detect important efficacy signals (Gelinas et al., 2017).
A screening log, also known as a pre-screening log, is a tool the research team uses to track why certain people are not eligible for a study. Clinical trial screening logs are constructed using the eligibility criteria listed in the research protocols. These requirements cover traits, including age, gender, past medical history and health status at the time, as well as specific criteria and risk factors relating to the disease being studied (See Figure 1). The screening log records the screening outcomes and each patient’s individual screening results. All screening efforts, including information from other sources (e.g. laboratory results), are entered into these screening logs. Screening logs make pinpointing the precise requirements that can impact participant enrolment easier. Many sponsors mandate that the site keep a screening log. As soon as a possible participant is found, the research team needs to note the status of each potential participant in the screening log (NIH National Center for Complementary and Integrative Health, n.d.).
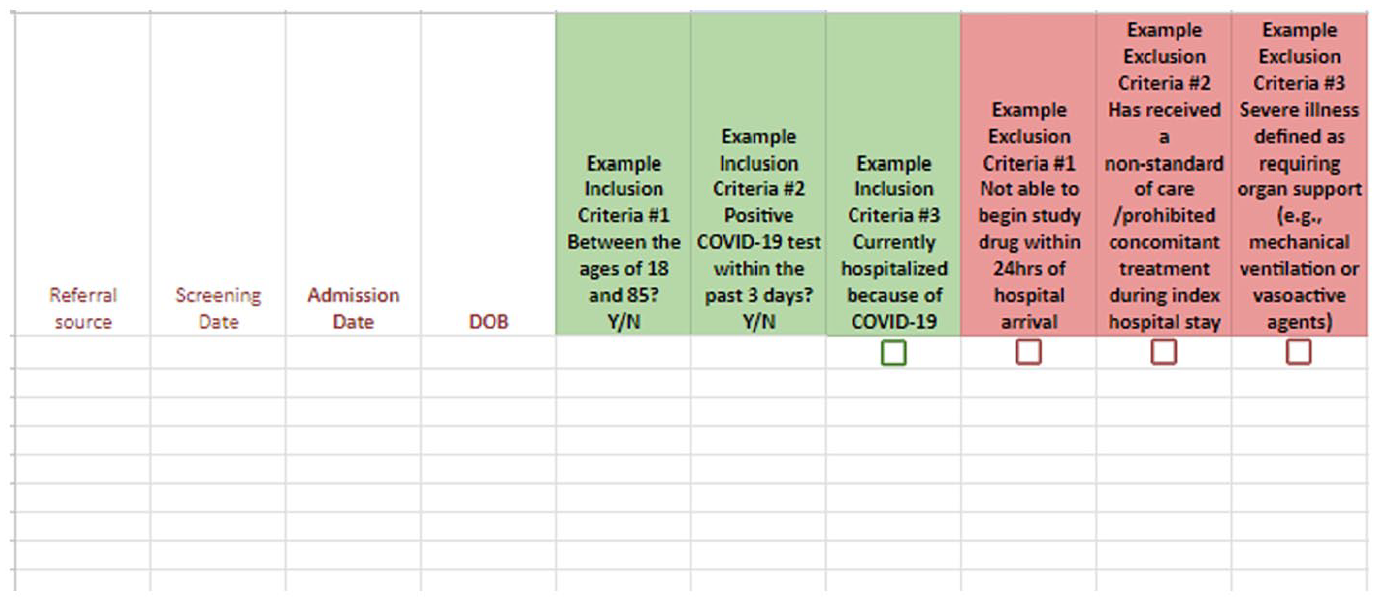
Example of a screening log used by study staff to determine patient eligibility.
Our clinical research institute (CRI) is part of a Level II Trauma Centre and Comprehensive Stroke Centre in the Midwest United States. Before the COVID-19 pandemic, the CRI had a small but robust clinical research department that managed clinical trials for stroke, cardiac, trauma and neonates, among other service lines. We worked hard to avoid trials with overlapping eligibility requirements. Our CRI began managing COVID-19 clinical studies in March 2020. The first COVID-19 studies at our site were mutually exclusive in their eligibility criteria. One trial enrolled seriously ill COVID-19 inpatients, whereas the other enrolled COVID-19 outpatients. We subsequently started another COVID-19 trial with moderately ill inpatients, so our populations did not overlap. As the treatment looked effective, the sponsor expanded participation to include participants hospitalised with severe COVID-19, creating an overlap between the two studies. This scenario occurred frequently during the COVID-19 pandemic. We needed to be able to adjust our objectives as the situation evolved. This paper describes how we used logic and clinical experience to evaluate and enrol patients ethically and efficiently while considering patient, sponsor and site priorities.
Screening and enrolment for competing trials prior to the pandemic
Gelinas et al. (2017) described four ways to manage competing clinical trials:
Approach 1. The site refuses to participate in competing trials.
Approach 2. Randomise which studies would be offered to participants on a first come/first serve basis.
Approach 3. The clinician decides which studies to offer participants based on resource availability, logistical concerns and time constraints.
Approach 4. The participant chooses from all studies they meet eligibility criteria for that is presented by the study staff.
Prior to COVID-19, in general, this last approach was the most common and considered the most ethically sound. Unless there were many potential participants (such as with oncology studies), the industry standard was for study sites to avoid competing eligibility criteria in their clinical trials. Allowing competition between studies reduces enrolment success (Gelinas et al., 2017), so it was in the site’s best interest not to have studies with overlapping criteria.
Before the pandemic, this research institute’s trial screening process could be spread over hours, days or weeks, depending on the study. The CRNs would screen for their studies by identifying a potential participant with an appropriate diagnosis, outcome or risk factor. In general, the number of available patients who met the criteria for any study was limited, and our enrolment goals might be one or two new participants a month. Patients may still have met the criteria for more than one clinical trial.
Introducing potential participants to a clinical trial and seeking their consent are lengthy processes. Our approach begins with summarising the trial objectives and its voluntary nature. If the potential participant is willing to hear more, a comprehensive version of the trial information is provided, and the consent document is reviewed. These conversations can last hours and be completed over several visits. Family members may be invited to provide input. Potential participants might be presented with one or more trials and are given plenty of time to consider the pros and cons of each study (Gelinas et al., 2017; Paquette et al., 2019). Study staff had ample time to answer questions.
The COVID-19 pivot
The COVID-19 pandemic created a new landscape for enrolment in clinical trials. Sathian et al. (2020) noted that enrolment in non-COVID-19 clinical trials dropped or stopped worldwide in early 2020, and new study start-ups were delayed or halted. Approaching potential participants became more difficult due to social distancing and the researchers’ hesitance to be exposed to a deadly virus. Remote approach and consent platforms were rare at the beginning of the pandemic. As the United States government put procedures in place to fast-track pandemic-related research, many clinical research sites pivoted to managing COVID-19 therapy clinical trials (Sathian et al., 2020).
Complicating the already complex recruitment procedure, potential participants for COVID-19 trials had less time to consider and ask questions because of strict eligibility criteria, such as the need for enrolment early in the disease process. This meant approaching participants in the earliest part of their hospitalisation, which could be a period of great stress. Patients are sicker and more anxious in the first days of their hospitalisation as they see multiple providers from the interdisciplinary team, have many therapies explained to them, and may be subjected to uncomfortable procedures. Presenting even one research study raises ethical issues by adding burdens to those already stressed. Outpatients were reluctant to come to the site or the hospital for fear of re-exposure and were unable to secure public or ride-share transport due to their positive COVID-19 status. These considerations for outpatient studies meant that patients were more likely to refuse participation in studies that required frequent in-person visits.
Screening and enrolment adaptions
Various amendments to our enrolment process were trialled. Our adaption process closely followed that of the aforementioned four approaches to management of enrolment for competing clinical trials described by Gelinas et al. (2017) (see Table 2).
Our approaches to match the industry standard methods of preventing overlapping criteria.
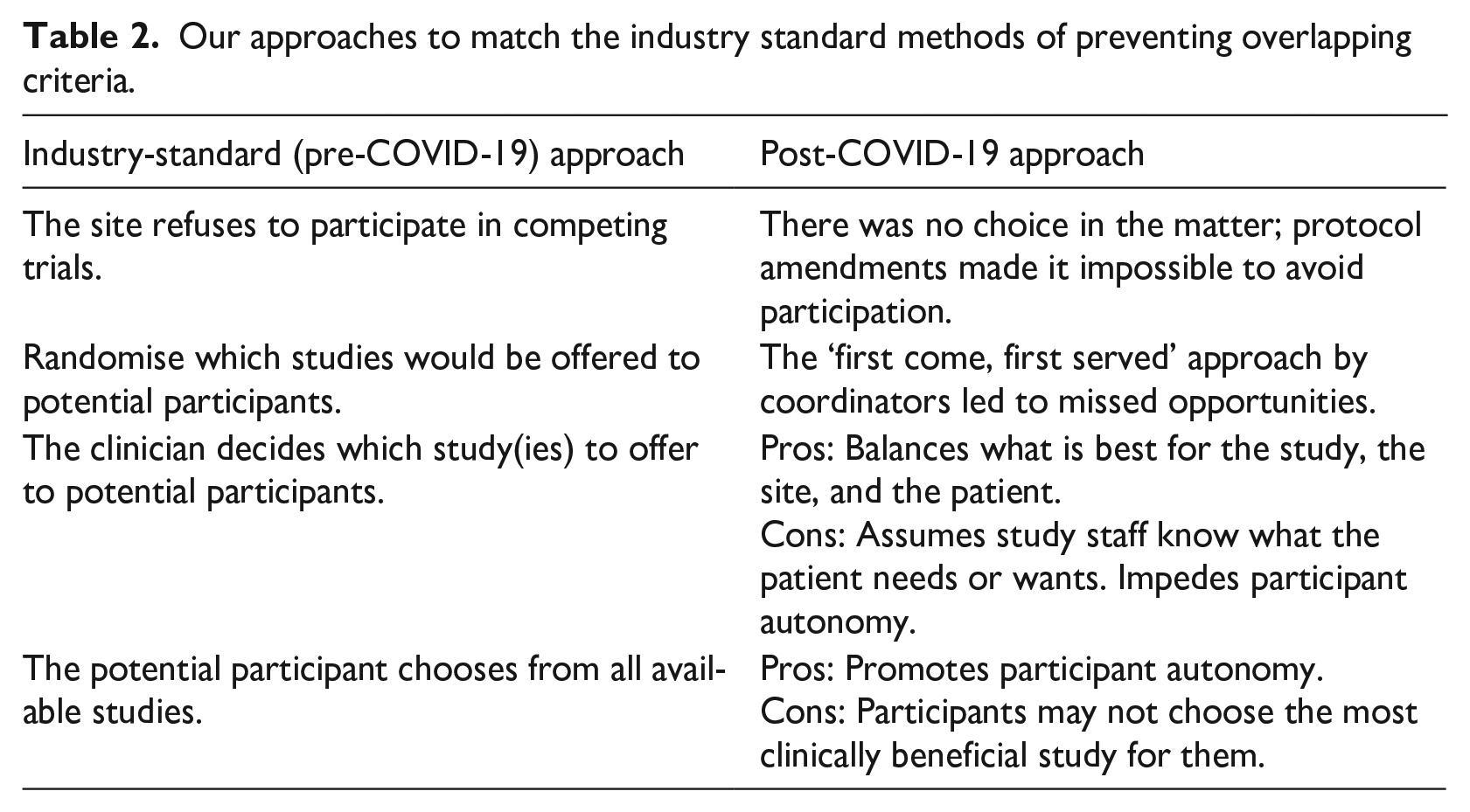
Approach 1: The site refuses to participate in competing trials
Our site informed sponsors about other COVID-19 clinical trials with competing enrolment requirements during pre-site selection visits. The sponsor seldom rejected our site because there were more than enough potential participants for any study. Indeed, competition could not always be anticipated with eligibility criteria that changed so often. One research study may initially recruit only moderately ill individuals, but protocol revisions may later allow severely ill people to enter. The trials would then become competitive if we already had a study enrolling seriously ill patients.
Approach 2: Randomised approach
Each of us was screening the same patient pool for our studies, and because eligibility requirements occasionally overlapped, a patient might be identified as a possible participant by two or more CRNs. Whichever CRN was screening a patient would talk to them without checking to see whether they also met the requirements for another study. We approached potential patients for our study, and if they refused, that was it. We would share that information with the other CRNs, and their names would be crossed off all lists so the patient would not get harassed with multiple approaches. Sometimes, this approach resulted in another CRN being blocked from approaching a patient for their study. Frustration ensued, especially if that patient might have been a better fit for a different study. We recognised that we were missing out on opportunities, so we changed our process to address it.
Approach 3: Clinician decides
A list of all positive COVID-19 tests in the regional hospital system was provided to the CRNs daily. The list sometimes included hundreds of names. The CRNs lacked the time to screen each participant for each study thoroughly. Before screening, coordinators would analyse the resources required to conduct the study and only screen for those we had the capacity for. For instance, a trial including nebulised treatment for COVID-19 required a negative pressure room. However, acquiring access to such a room became increasingly difficult as the incidence of COVID-19 increased, and all available space was shifted to inpatient use. If a negative pressure space was unavailable, a patient could not be offered participation in the trial. For other research trials, coordinators may have chosen which study to contact a patient about based on considerations such as the distance the patient would travel to the study site. Some outpatient trials required visits every other day; hence, giving such a study to a participant who lives 100 miles away risks loss-to-follow-up. All of this was done to reduce the number of individuals to screen and approach to a manageable level. We would place the patients on the screening log of the study for which they had the highest likelihood of acceptance and then approach them.
Approach 4: The Participant chooses from all eligible studies
We tried offering multiple trials to inpatients and letting the patient decide. Sometimes, they were able to consider the studies carefully and make a choice about which one they wanted. Other times, the volume of information presented probably overwhelmed them, and they refused to participate in any study. Complexity confuses and increases the likelihood of a flat refusal (Ali, 2002; Kaur et al., 2014). Would we have had more success if we had offered those patients just one study rather than complicating their decision by presenting more than one study?
We also tried presenting all available trials to outpatients. They almost always chose the study with the best chance of receiving the investigational product and requiring the least effort (and the fewest blood collections). The participant may find certain options appealing, but it is essential to consider that they may not be the most beneficial or could potentially pose a higher risk for them. Fewer blood draws mean less safety monitoring of the patient’s condition. Novel therapy studies almost always require more from the participant. When we offer two studies to patients, one open-label and one with 1:1 randomisation to investigational product or placebo, it is easy to see which study would have more successful enrolment. When one study required visits every other day for a month and another required weekly phone calls, one can guess which study sounded more attractive to participants. Sometimes, offering all studies to all participants may not be feasible, even if they meet the criteria.
Merging approaches 3 and 4
Our efforts resulted in a new way to screen and approach potential participants (see Figure 2). We dedicated a screening person every day to screen for COVID-19 studies. We had a second person verify the eligibility criteria. Any red flags like potential non-compliance, distance or drug use were discussed between the lead CRNs. We posed questions such as: Is this study going to be a more significant risk for this patient than other patients? Is the patient at a higher risk of disease progression based on comorbidity? Is it possible that one study would be more beneficial to them? Then we would have a conversation about logistics: Are tests going to need to be repeated that would delay enrolment? Can we provide the treatment and meet the requirements of the study according to the protocol? We notified the principal investigator and had them review eligibility to ensure they agreed with our assessments. If this were an outpatient, we would read their providers’ notes and make sure nothing had been documented that might concern us about their participation.
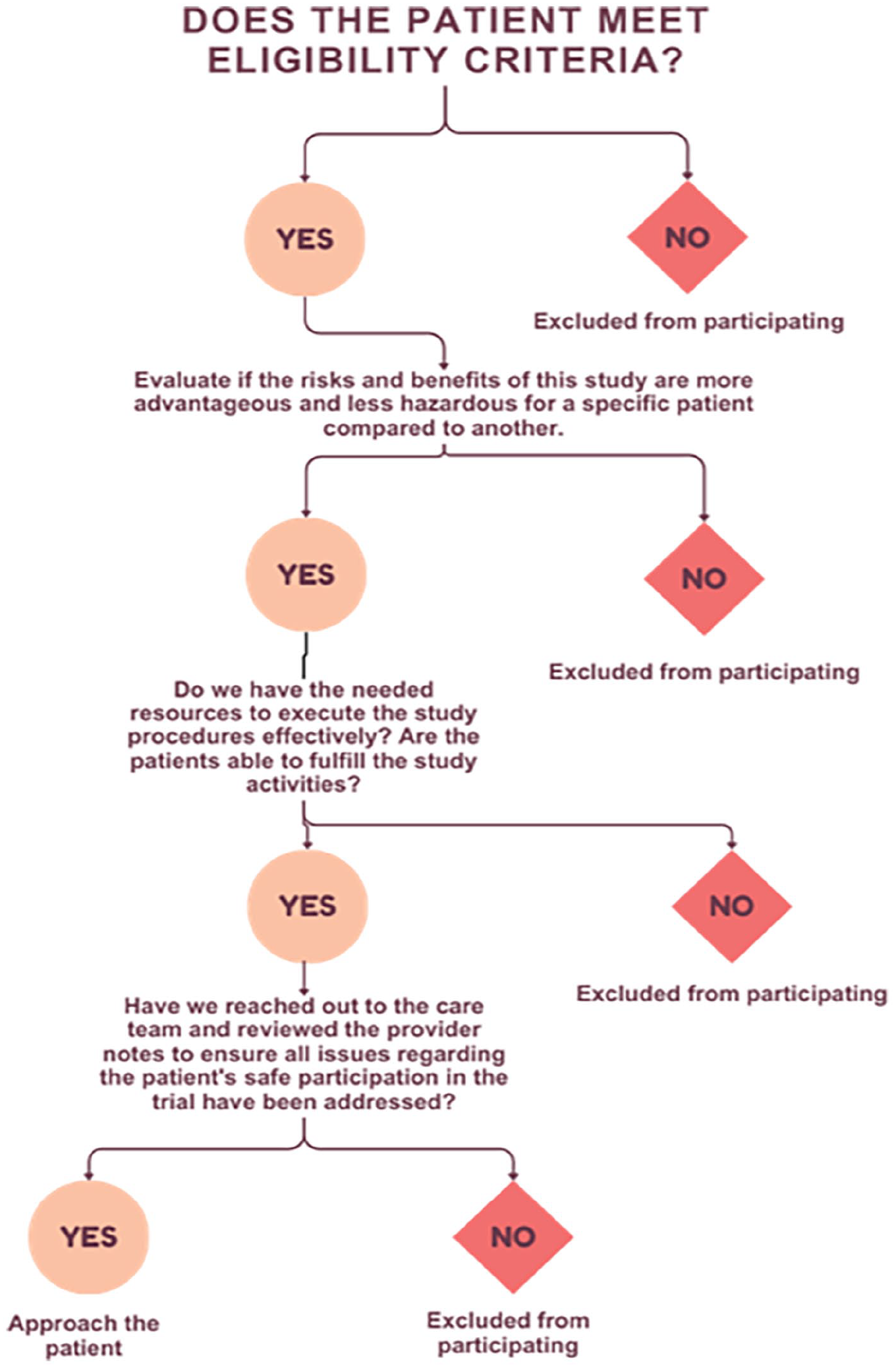
Screening workflow: Beginning with a chart review, CRNs rapidly screen a patient for all COVID-19 studies. After determining eligibility, the CRN considers the resources needed to complete the study requirements. For inpatients, the care team is also consulted regarding any concerns about the patient’s suitability. For outpatients, provider notes are extensively reviewed to determine whether there are additional concerns. Finally, the patient is approached and offered any studies that match their situation.
For hospitalised patients, we approached the bedside care team for their assessment of the patient, making sure they did not have any concerns about the patient’s involvement. We had not yet spoken to the patient at this point, but we knew a lot about them, such as the specific concerns for their disease state and whether one of our studies might address them. We approached the patient and invited family members to join the conversation. We presented the study to the patient and highlighted the potential risks and benefits for that person rather than presenting a random and disconnected study. We could not guarantee any benefit, but we could explain relevant potential benefits and address concerns that that patient might have. Patient autonomy is best supported by discussing their health concerns, specific risks and benefits from the studies and acting as interpreters and advisors (Jansen et al., 2021; Kaur et al., 2014). By bringing in all stakeholders, we educated ourselves sufficiently about the patient’s unique situation for an in-depth conversation. If all stakeholders had genuine uncertainty about the superiority of one trial over another, we could offer all trials to the participants and let them decide. Clinical expertise and thoughtful consideration of a patient’s unique situation may lead to the patient being offered the best trial or trials for them.
Discussion
The above method has given us a way to help decide which patient gets offered which study or studies. It has required a balance between our priorities as clinicians and researchers. However, the process did involve us making assumptions about and decisions on behalf of the patients. The patient’s decision-making power was reduced to a single yes or no answer. Conscientious enrolment in clinical trials requires collaboration with various stakeholders, primarily focussing on patient rights and safety. However, it is acknowledged that ethical tensions and competing priorities need to be carefully addressed.
While it is not a legal requirement to disclose all clinical trials available to patients, can we say that we have obtained fully informed consent if we have not informed the participant of all other options, including other clinical trials? Limiting the number and type of trials offered may impede the patient’s autonomous decision-making. The principle of autonomy is a fundamental concept in medical ethics, often highlighted in documents like the Belmont Report (Fischer, 2006). It emphasises the importance of respecting individuals’ right to make their own decisions regarding participation in research. In keeping with these ethical principles, we do our best to notify patients if they qualify for multiple studies and explain the selection process for the specific ones chosen for them. The patients can acquire additional information on any of the studies.
What if they choose a study that might pose a higher risk for them? While all clinical trials must have a certain balance between the proposed risks and benefits, patients are unique, and certain trials may hold greater risk for one participant than another. Researchers may overlook cumulative risks when weighing an individual’s potential risk versus benefit. For example, an MRI or blood draw might not be very risky on its own, but several of these procedures done throughout a study might put participants at more than minimal risk. It is crucial to consider how interventions as a whole affect participant welfare and safety (Wendler and Miller, 2007). Medical practitioners must act in the patient’s best interest while protecting their autonomy rights (World Medical Association, 2013). While the site’s responsibility to prioritise patient welfare may clash with institutional goals for meeting enrolment targets and financial considerations, as outlined in documents like the Declaration of Helsinki (Fischer, 2006), the principle of beneficence underscores the obligation to maximise benefits and minimise harm to research participants.
Clinical trial ethics also requires consideration of equipoise (Rabinstein et al., 2016). Additional considerations should only be made if there is equipoise − a genuine state of uncertainty − regarding the welfare of the patient and the benefits to society (Rabinstein et al., 2016; Wendler and Miller, 2007). For instance, in a clinical trial assessing two potential treatments for a life-threatening illness, a state of equipoise may exist concerning the welfare of individual patients and the broader societal benefits. In such a case, the ethical analysis would involve considering factors such as the potential risks and benefits for individual patients, the impact on public health, and the allocation of limited resources (Wendler and Miller, 2007). This would require transparent and justified decision-making to ensure the conflicting ethical principles are carefully addressed to promote patient welfare and societal benefits. Transparency and justification of decisions are integral to ethical conduct in research, as emphasised in various guidelines, including the World Medical Association’s Declaration of Helsinki (World Medical Association, 2024).
We suggest that these procedures provide a framework for researchers to navigate complex ethical dilemmas and make informed decisions. By considering the potential impact on individual patients and society, researchers can ensure that their actions align with ethical principles and contribute to advancing medical knowledge. (See Figure 3)
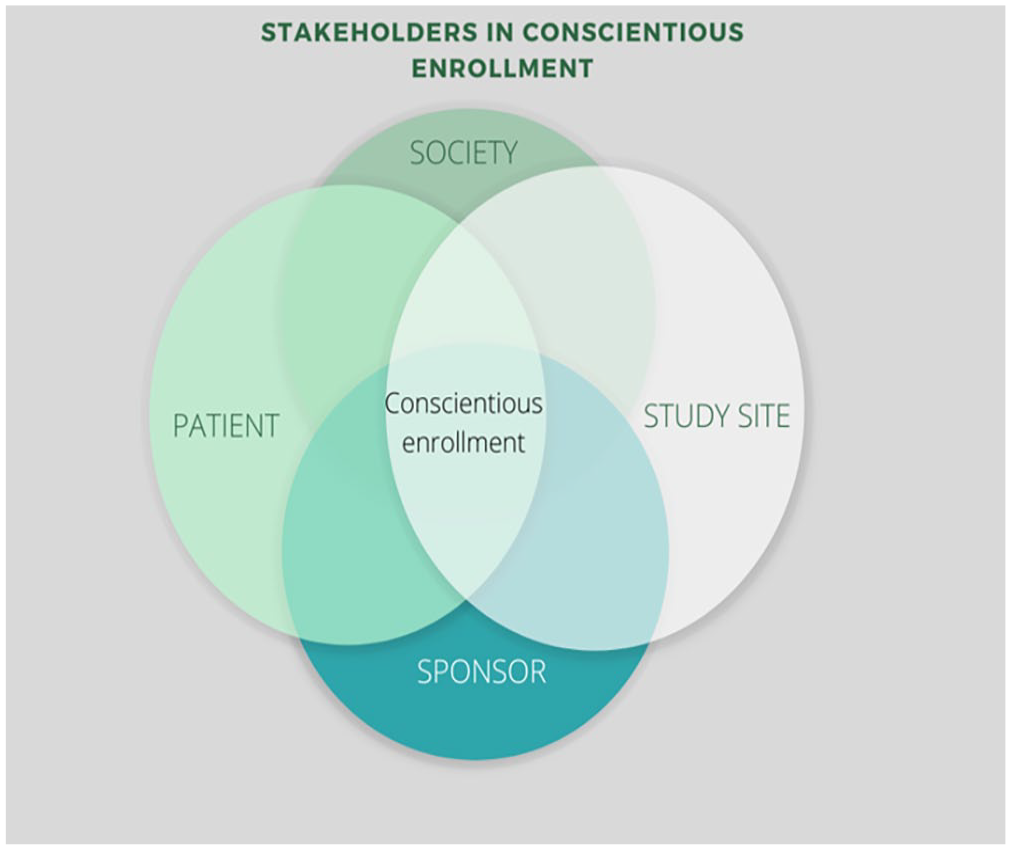
Conscientious enrolment requires considering the needs and priorities of all stakeholders.
While the CRI’s conscientious screening and enrolment process is a good start, it has limitations. Since patients’ preferences and needs change over time, it may be difficult to accurately determine what they want and feel is best for them. Additionally, the provider’s beliefs and the institution’s goals might not always align with what is best for the patient. While considering the sponsor’s and society’s interests is essential, it may introduce biases and conflicts of interest that compromise the clinical trial. Thus, while this process aims to consider multiple perspectives, its limitations must be acknowledged and managed.
Footnotes
Authors’ Note
MA is a board-certified clinical research nurse directly involved in subject recruitment, obtaining consent, clinical trial management and stakeholder engagement.
Authors’ contributions
MA was the primary author. SM contributed significantly to the writing and revision of the manuscript. KM contributed significantly to the writing and revision of the manuscript. AB contributed significantly to the writing and revision of the manuscript. JB guided the project and contributed significantly to the conception, writing, and revision of the manuscript.
Availability of data and materials
Not applicable
Consent for publication
Not applicable
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.
Ethics approval and consent to participate
No human participants, human data, or human tissue used. Approval waived.