
Abstract
Simulated standardized patients (SPs) are trained individuals who pose incognito as people seeking treatment in a health care setting. With the method’s increasing use and popularity, we propose some standards to adapt the method to contextual considerations of feasibility, and we discuss current issues with the SP method and the experience of consent and ethical research in international SP studies. Since a foundational discussion of the research ethics of the method was published in 2012, a growing number of studies have implemented this method to collect data on the quality of care in a variety of settings around the world. We draw from that experience to provide empirical foundations for a popular approach to ethical approval of such studies in the United States and Canada, which has been to obtain a waiver of informed consent from the health care providers who are the subjects of the research. However, the majority of studies to date have evaluated quality of care outside the U.S., requiring additional ethical consideration when partnering with international institutions. We discuss these considerations in the context of a case study from a completed SP study in South Africa, where informed consent is constitutionally protected.
Introduction
There is an urgent need to conduct quality of health care research in such a way that ethical conduct is centered and clearly explainable to partners and stakeholders. One research method that has recently become popular—simulated standardized patients (SPs)—has been shown to resolve problems of social desirability bias and observation bias by collecting key counterfactual data with high precision (Das et al., 2022). The SP approach is a highly effective method that observes the treatment of the exact patients, settings, and providers that researchers are interested in (Kwan et al., 2019). By allowing researchers to generate high-quality data in this fashion, the method resolves the Big Data Paradox that often occurs in administrative health care data: “The bigger the data, the surer we fool ourselves” (Bradley et al., 2021; Meng, 2018). However, because the SP method typically involves covert audits of health care provider performance—often with a waiver of informed consent—there are serious ethical considerations in the method.
SPs are trained staff enumerators who pose incognito as actual patients seeking care in a health care provider’s place of practice in order to measure the quality of care delivered by those providers (King et al., 2019). Individual SPs are hired from local populations and trained to portray a specific predetermined medical condition to health care providers, then to recall accurately and in great detail the actions of the provider. After completing each interaction, the SPs debrief with a manager and complete a structured questionnaire detailing the consultation and the actions the provider took. This questionnaire is the only data collection; the method does not collect protected health information about any research subjects.
Compared to other quality of care measures, SP studies provide an unbiased measure of many dimensions of care. The exact needs of the patient simulated in the scenario are known to the researchers, and the researchers select which providers are observed (Kwan et al., 2019). For example, antibiotics are required to treat some conditions; for others they are contraindicated. The only way to evaluate the appropriateness of any observed pattern of antibiotic use is to know the condition of the patients. Administrative data is unsuitable to this purpose because incorrectly diagnosed or treated patients cannot be detected; incorrect diagnoses are generally indistinguishable from correct diagnoses. Follow-up interviews with patients or providers provide little additional information to distinguish patient needs. Even complete administrative data may not be able to determine justifiable variations in antibiotic use because of unobservable population needs, patient mix, and sorting (Sulis et al., 2020). By contrast, SP data can quantify overuse as well as underuse of such services.
Other technical elements of the SP method make it highly attractive for research purposes (Kwan et al., 2019). The ability to deliver identically scripted case presentations to a selected provider sample allows targeted research questions about quality of care to be addressed. It removes confounding issues due to patient selection of providers depending on the (self-)perceived severity of their case, as well as confounding due to barriers to access. Rigorous training of enumerators ensures that SP data is complete and high-quality, and the “blinded” design in which providers do not know they are treating an SP ensures there are no Hawthorne effects or desirability biases in the observations. Due to the high data quality and relatively small sample sizes required, the SP method has become an essential research tool for measuring quality of care, as well as providing causal answers to questions about differences in care, quality improvement initiatives, and patient presentation (Das et al., 2012, 2016a, 2016b).
Many SP studies have been performed with IRB-approved waivers of consent by providers, justified by a combination of minimal risk and protecting research integrity, sometimes conditional on informing research subjects after research activities were completed. The purpose of SP research has been to reach actionable conclusions that other methods cannot achieve about the structure and function of whole health systems for the benefit of partners and policymakers. The accuracy of these aggregate measures is essential to the public good element of such research—especially providing causal conclusions about crucial topics like gender discrimination, health insurance programs, professional training and medical education, and the like; recent developments in causal and experimental study design using SPs continue to underscore this strength (Kovacs et al., 2022; Kwan, 2020).
Issues around the ethics of using such waivers have been discussed most prominently by Rhodes and Miller (2012), and this article engages with that discussion through empirical evidence collected in the years since then (Rhodes and Miller, 2012). Over that time, we and other researchers have conducted a wide range of SP studies, both with and without waivers of informed consent by providers (Das et al., 2022; Wiseman et al., 2019). Here, we report empirical experiences demonstrating that SP research generally meets U.S. federal requirements for waivers of informed consent with deception in data collection. Studies that demonstrate the necessity of deception as well as document minimal risk to research subjects (health care providers), as well as to SPs themselves and to other patients and individuals, are suitable for waiver requests. A range of evidence supports the assertion that under these conditions, such a waiver is essential to the practical execution of the research, and that requiring informed consent can jeopardize research findings and be harmful to public health by introducing large, unknown measurement biases.
However, these needs alone do not by themselves justify a waiver of informed consent. In theory, a research team could approach providers in advance and complete a consent protocol informing providers that they will be observed incognito at some point in the future. We address case studies of SP research in South Africa, reviewing the complexities of waivers of consent in the post-apartheid context (Burger and Christian, 2020) and discussing implications for the scientific validity of SP studies. We report in detail the experience of one study in South Africa undertaken with an explicit informed consent protocol (Christian et al., 2018; Salomon et al., 2022). The results of this consent process—namely, high dropout rates subject to unknown biases—support the conclusion that consent waivers are generally necessary to avoid compromising the scientific validity of SP studies. Such waivers can, depending on context, be conditioned on higher-level approvals such as administrators or governments, follow-ups with providers who were visited, or other appropriate actions. Finally, we reflect on the language of the South African right to informed consent, and conclude with a discussion of ways to foster responsible research partnerships that consider international and post-colonial contexts without compromising the strengths of the SP method (Khan et al., 2022).
Waivers of informed consent in SP studies governed by U.S. regulations: Empirical experiences
It is well-established that provider performance generally falls short of demonstrated knowledge and that direct observation is essential to measure quality (Mohanan et al., 2015). At first glance, an SP audit approach often appears to simply be a laborious and limited method of collecting data on quality of care in contexts where rich administrative data is unavailable. Studies and reviews in the quality of care literature often make this assumption implicitly through research questions such as “compar[ing] and contrast[ing] the advantages and disadvantages of each method” in measuring quality (Aujla et al., 2021).
However, even complete and accurate administrative data would be unable to answer research questions which are approached by SP studies, because administrative data has an unsuitable data-generative process. First, it is impossible to know the underlying medical conditions of care-seekers without extensive and unethical approaches such as mass surveillance and testing. Second, it is impossible to remove confounders due to patient selection of providers depending on the perceived severity of their case, as well as confounding due to barriers to access such as prices, geography, perceptions of providers, and so on. Third, it is essential that research teams are able to observe appropriate counterfactual interactions with different providers or patients. By pre-determining the underlying condition, the demographic background of the individual, and the complete responses to questions that the provider might ask, as well as controlling the selection of the provider sample, research teams have used the SP method to determine the quality of care across study populations of providers (King et al., 2019).
To effectively implement this method, research teams have frequently requested approval for deception of health care providers in combination with a waiver of informed consent by them. This requirement is detailed in 45 CFR § 46.116, “General requirements for informed consent.” U.S. regulation provides that: “An IRB may waive the requirement to obtain informed consent for research under paragraphs (a) through (c) of this section, provided the IRB satisfies the requirements of paragraph (f)(3) of this section.” There are five requirements for such a waiver, and we address each in turn here with empirical details from prior work.
The research involves no more than minimal risk to the subjects
Minimal risk is defined in 45 CFR § 46.102 as a study in which “the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.” Correspondingly, the extent of providers’ participation in an SP study is performing their usual professional duties with a care-seeking individual—and, unlike in an ordinary patient interaction, SPs are guaranteed to not expose providers to potential contagion. Individuals are trained to be fully compliant with provider instructions to the extent that it is safe; therefore, the provider’s routine activities and choices determine almost every facet of the interaction.
One study reported results from a sample of consenting providers who were later debriefed on their participation: “None of the 93 providers who completed the detection survey voiced concern that participation in the study adversely affected them. Additionally, no financial losses were incurred by providers because the standardized patients paid normal fees to receive services, and there were no added inconveniences to the provider because standardized patients were trained to immediately step aside if an emergency occurred in the clinic that demanded the provider’s attention” (Das et al., 2015).
A general source of potential risk to providers during SP studies is the disclosure of personally-identifying information (PII) during or after the study. The authors have often been asked whether any action is taken by researchers when inappropriate or even dangerous medical practices are observed in SP data, such as disclosing such actions to employers or regulators. While such action might be appropriate if a private individual were to observe such an action, it is never acceptable to disclose personally-identifying information about research subjects—here, providers—under U.S. regulations. In accordance with such regulations and in our experience with all governing IRBs, the expectation is that SP projects undertake standard data protection practices, including: encryption of all data in transit and at rest; de-identification of all data before publication or release; and publication of appropriately aggregated statistics when individual measures have the potential to be identifying. However, SP data does not generally warrant the additional security and privacy measures required for data that fall under HIPAA or FERPA regulations in the U.S. or higher.
Other people may also be affected by SP research activity—namely, the SPs themselves, non-SP care-seekers, bystanders such as family members, and non-medical staff. None of these people are research subjects; but research teams must still take seriously their well-being and safety. For example, SPs could cause delays in providers’ ability to attend to other patients. “This time burden could be substantial if the delay affected seriously ill patients; however, in those circumstances, SPs were trained to allow others to bypass the queue and see the doctor immediately [. . .]; this situation did not arise during fieldwork” (Daniels et al., 2017). There is no reason to believe that the time spent seeing SPs in non-emergency situations poses more than a minimal burden to other care-seekers. Studies typically reported wait times of 30 minutes to 2 hours, which might potentially be extended by the duration of a typical interaction, usually 5–10 minutes.
SPs themselves could also be placed in danger. The most common risk observed has been that a provider would want to administer medication during the interaction, with injections being common in some settings. Research teams have developed extensive safety protocols for SPs to refuse a variety of such attempts and, with training, risk avoidance has been successful (Kwan et al., 2019). The next most common risk has been that a provider would want to conduct an invasive exam or medical test. Depending on the type of procedure, this may or may not be acceptable. An external thermometer measurement or blood pressure reading could generally be acceptable; a sterile finger prick for a rapid blood diagnostic may be acceptable if required for the research and agreed by the SP. Procedures such as X-rays, EKGs, or genital exams must be avoided and have caused research protocols to be revised or abandoned if providers repeatedly requested them (unpublished). We again emphasize that the SPs themselves are voluntary staff and are trained to avoid any medical or social risk, in addition to having the ability to communicate with managerial teams in case of emergency.
To further protect SPs, researchers have implemented extensive protections to avoid them being detected or suspected as SPs. As one study reported, “To assess detection rates, each provider was administered a structured questionnaire within 2 weeks of the completion of fieldwork asking whether an SP had visited the clinic, and, if so, the characteristics of the SP. . . detection rate for the SPs was zero in this study.” Subsequent studies have all reported similarly low detection rates, ranging from 0% to 4% (Boffa et al., 2021; Daniels et al., 2017). However, detection risks may vary widely depending on factors like the size and type of communities in which studies are taking place, on the typical processes of communication and introduction between providers and patients, and on other considerations like national ID numbers and insurance. These must be carefully understood by research teams in the development phase of any SP study.
The research could not practicably be carried out without the requested waiver or alteration
To answer quality of care research questions, research teams must be able to construct scientifically valid samples of providers appropriate to specific hypotheses. It may be feasible in very small samples to attempt to obtain informed consent from every provider for SP studies while preserving the methodological integrity on an individual basis, for example by informing them that they will be visited by an SP in some indefinite amount of time, without disclosing the specific condition the SP will present with. One immediate issue is that there is no guarantee providers will consent. This is never a sufficient reason on its own to justify a waiver, but plausible patterns of non-consent (such as by low-quality providers or specific demographic groups) could easily undermine the scientific validity of SP research conclusions and potentially do harm to the health systems the research is meant to inform.
Explicit informed consent is both logistically impractical and methodologically inappropriate in many SP studies. The core reason for this is that, in a large number of common situations, it is not possible in advance to determine the identity of the provider(s) who will attend to the SPs. The SPs could be directed to any number of available providers by a triage team; handled by a substitute due to absence of a specific provider; treated by an auxiliary or visiting member of a medical team not listed as a primary care provider; or assisted by a community health worker or other individual, to name a few common situations. One study reported that in 64% of interactions in rural India, SPs were treated in public clinics by someone who was not the named provider, but who was serving as a substitute at the time of the visit (Das et al., 2016a). Partly as a result, many studies draw samples at the level of the health facility, not the individual provider, since this is often how patients seek and receive care before being triaged to a specific provider.
Allowance for this indeterminacy of potential research subjects is desirable because it reflects the true process of care-seeking. Even if informed consent could be guaranteed for all relevant individuals, however, it is highly impractical for researchers to identify, contact, and consent all the people who might become subjects of the research in a timely and complete manner. It is also often impossible for the SPs to identify by name the individuals who do in fact attend to them, making identity matching and consent validation infeasible as well. As a result, for all practical purposes, it is not possible to conduct SP research with informed consent from every individual who might interact with the SP and have their actions recorded in the course of the study. These conditions together make much SP research scientifically invalid without a waiver of informed consent.
If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format
In order to conduct the data collection and analyze the results of the study, it is essential to be able to match the outcomes of interactions against the research population. Therefore, personally-identifying information for facilities and/or providers is generally recorded and maintained as a securely stored database of names and locations. This process is handled identically to any statistical survey collecting personally-identifying information or other sensitive data types; the practical considerations of these protocols are well-established and followed.
Crucially, however, no non-SP patients are ever observed, and no biospecimens or protected health information are ever collected from research subjects. In fact, the use of the SP method eliminates the need to collect such sensitive and protected information. Questions about the appropriateness of care for individuals cannot be answered on a population basis by observing providers’ treatment of patients without knowing the true health status of the individuals. Since diagnoses are often missing or incorrect in administrative data, health records and the like cannot typically be used for these questions. Instead, researchers would have to engage in approaches such as testing every care-seeker for a range of rare conditions after observing their interaction with the provider. Such an approach is highly unlikely to be ethical; in addition, it is laborious, invasive, and impractical, requiring the research team to handle both biospecimens and protected health information.
The waiver or alteration will not adversely affect the rights and welfare of the subjects
In their essential analysis, Rhodes and Miller (2012) write: “It might seem impossible for simulated patient studies to satisfy [this] condition, as they do infringe on rights not to be deceived and to be free of research interventions without consent” (Rhodes and Miller, 2012). In a similar analysis of deception in research, O’Neil and Miller (2009) write: “Common morality recognizes that rights may be permissibly infringed for sufficiently good reasons” (O’Neil and Miller, 2009). In the case of SPs, we rely on the judgment of IRBs that the value of the research for the public good outweighs the temporary infringement (not violation) of providers’ rights to refuse participation or deception as a scientific subject. O’Neil and Miller continue, “Accordingly, the question of whether obtaining authorization for deception is practicable is difficult to answer without recourse to systematic empirical investigation of the reliability of authorized deception.” We address this in the case study that follows, demonstrating that the risks of informed or authorized consent or deception in SP research are likely “so grave that they would undermine the research.”
As for welfare, health care providers are never subjected to any conditions other than those they routinely expect as part of their regular professional duties. Their identities are never revealed to any third party, nor retained beyond the information needed for scientifically valid sample construction and data interpretation. Research subjects therefore face no potential consequences of any form as a result of their inclusion in the study, either positive or negative, other than their usual compensation for time and work. In one study, it was reported that “[t]he burden on providers was the time spent with SPs, which averaged 7.2min per interaction (95% CI: 6.1 to 8.2) [. . .]” (Daniels et al., 2017).
That study further noted that private providers are always compensated for their time and all prescribed medication at their regular rates, so the uncompensated burden on providers should be zero where providers determine their own fees. SPs are trained to be completely compliant with provider instructions up to the limits of personal safety, purchasing all services recommended by the provider. In not-for-profit facilities such as the public sector, medical services may free or subsidized, and therefore provider time spent is a true additional burden on the provider and any medications given for free are a true burden on the payer. However, for outpatient interactions these costs are typically minimal, especially compared with the salary of the providers, which are already fixed and budgeted and pose no additional burden from the SP study. When third-party payers such as insurance are involved, research teams have in the past arranged with those firms to purchase and/or compensate expenses on a case-by-case basis; each research team must make these determinations in context.
Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation
In SP studies that seek a waiver of informed consent, it is often the case that notifying health facilities or providers that they are the subjects of active SP studies can in fact create risks. These risks are to (a) the SPs and (b) to the general public. This is because a large share of individuals suspected of being SPs by informed providers have fact been non-SP care-seekers. Detection of actual SPs is far less common: “Providers claimed to have detected an SP in nine instances, but on further elicitation of the characteristics and presentations of the suspected cases, there was no match between the SPs we had sent and the presentation of the patient that the provider suspected to be an SP” (Daniels et al., 2017). Therefore, notifying providers about the SP study except in the general sense of research publication could change their behavior outside the study and potentially negatively affect the welfare of the general public, which would be an inappropriate consequence.
In exceptional cases where unforeseen adverse consequences might arise, for example in a serious emergency or in case of detection, SPs are trained to reveal themselves and the study to the provider with a scripted statement. In such cases, the subjects are then provided relevant information about the study and the interaction is typically abandoned as incomplete. In completed interactions, by contrast, it is often impractical to retrospectively identify the exact individual who participated in the SP study, since health care providers may not be specifically targeted and any staff member could attend to each case. In most non-emergency cases, debriefing the subject is therefore impractical, and in fact only poses a new risk in the study. Word can spread quickly among medical communities, and descriptions of “suspicious” individuals can circulate, potentially impacting both the quality of medical care for the general public as well as decreasing the value of the study for improving public health.
Case study of informed consent: South Africa
We now discuss two published studies that have been carried out using the SP methodology in South Africa. The first evaluated TB management in primary care clinics in the public sector in two South African cities (Cape Town and Buffalo City), for which a waiver of consent was sought and approved by an institutional ethics board in 2014 (Christian et al., 2018). The second study looked at a similar question among private general practitioners (GPs) in Cape Town and eThekwini, for which a waiver of informed consent was not sought from institutional ethics boards in the 2017 application (Boffa et al., 2021). In the latter study, research assistants were trained to seek informed consent by visiting the practices of GPs registered with the Health Professions Council of South Africa, in selected communities.
Eligible GPs were informed that they would receive up to three SP cases with “a set of common symptoms” over a 6-month period. All SPs would pay regular GP rates, wait in queues as any other patient would, and step aside for any medical emergency. Consenting physicians were asked in advance to document any patients they thought may have been SPs during the study period, recording as much detail as possible, including presenting complaint, date of visit, gender of patient, and reason for suspecting the patient was an actor, which enabled researchers to estimate detection based on follow-up phone calls. Exit interviews with SPs included questions about whether they thought detection occurred during a doctor’s visit (overtly or suspected) and whether the SP made errors in their case presentation as an additional data check.
Ethical approval
In general, the rules for seeking a waiver of informed consent in South Africa are much more stringent than in the U.S. (Cleaton-Jones and Wassenaar, 2010). The South African constitution, passed into law in 1996, includes the right to informed consent for medical or scientific experiments. Department of Health guidelines clarify that “The term ‘experiments’ originates from Article 7 of the International Covenant on Civil and Political Rights – UN 1966 and echoes the Nuremberg Code; in the constitutional context, it is intended to mean ‘research’” (South Africa Department of Health, 2015). The researchers from the two South African SP studies referred to herein approached the matter with equal care, but to different ends in part due to the population of providers being studied. Christian et al. worked closely with respective Health Impact Assessment and Research Units, provincial TB Control Programs, Department of Health District Management teams, and the Chair and bioethicist of the institutional ethics committee at Stellenbosch University (HS1096/2014-REC) to seek (and receive) a waiver of informed consent for SP data collection in public sector clinics for the purposes of measuring adherence to national care guidelines.
By contrast, Boffa et al. opted not to seek a waiver of informed consent, ensuring that individual private providers were afforded their right to decline to participate as required by South African research ethics guidelines. While client needs may differ in the public sector depending on community, care decisions made by private sector practitioners can further differ between high- and low-income communities due to differences in socioeconomic factors and disease profiles driven by dynamics of cost, insurance coverage, and the like. Ethical approval with informed consent was granted by the Human Sciences Research Council in South Africa (ref 2/18/10/17) and McGill University Health Centre in Canada (2018-4390).
Findings
In the Boffa et al. study, 13 communities in Cape Town and seven in eThekwini were selected with a total of 641 eligible providers identified through online listings. Among 385 GPs who responded to consent requests, 164 declined (42%). Patterns of consent varied across study sites; in one site, 58% of women declined to participate, while in the other, only 17% of women declined. Participation among men was approximately 65% in both sites. Racial breakdowns were not reported. Figure 1 illustrates the full sample consent process; the demographic statistics of those who did not participate were often not recorded.
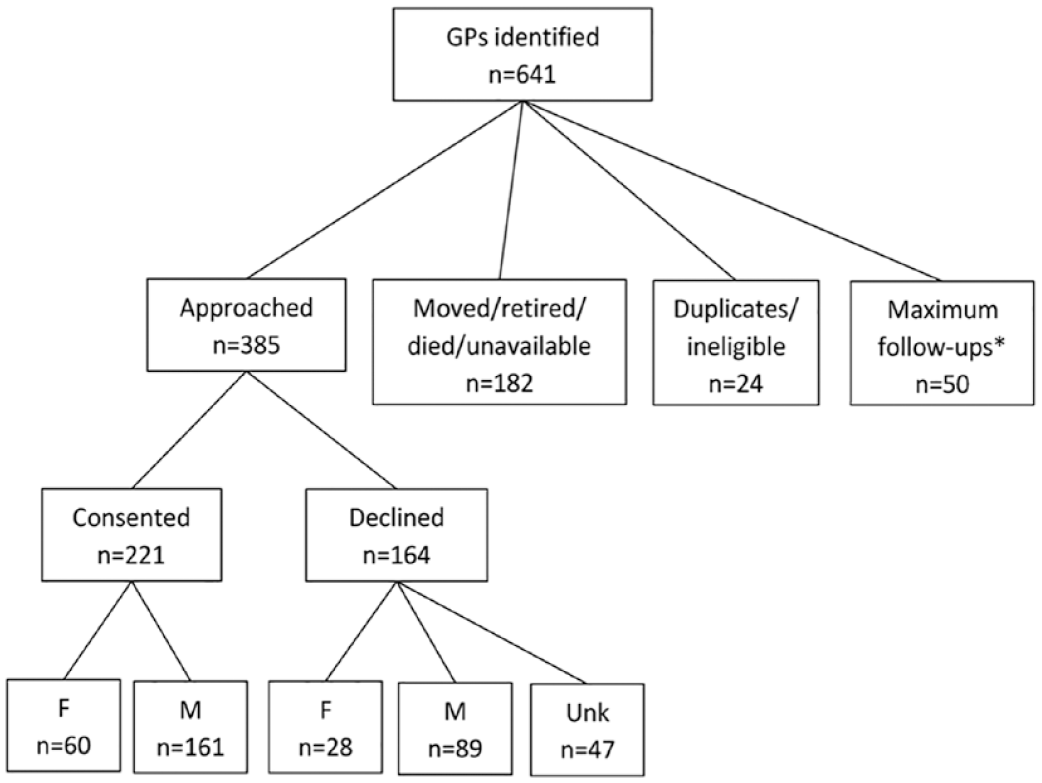
Informed consent among private providers in Cape Town and eThekwini, South Africa.
During the process of seeking informed consent, it was very common for a long delay. For example, providers were occasionally unwilling to give a firm rejection. Instead, as the consent team reported, providers often said they were “going to think about it” and would continue doing so in each follow-up attempt. As a result, the team implemented a five-attempt maximum follow-up cutoff, after which the provider would be excluded from the study. Of 435 informed consent attempts, 50 (11%) of eligible providers fell into this category.
During SP interactions, there were several issues encountered characteristic of the challenges of working with informed consent. When SPs were sent out, it was not uncommon that they would encounter a temporary replacement provider. SPs were prepared with physical descriptions of the consented practitioner (“the tall one,” “the one with a mustache”), to differentiate from other GPs in the practice or a locum (substitute) that may be visiting, which was provided to SPs in their assignment sheets. Scripts were prepared for SPs to abandon interactions with a non-consented provider when the SP encountered the wrong individual in the consulting room, such as to pretend their mobile phone rings and to say that there is an emergency.
In all cases—although it is unusual for non-SP patients—SPs would clarify the provider name and, if an incorrect provider was attempting to attend to them, indicate a strong preference to see the selected provider when they return, or to wait until that provider became available if they are there. SPs would always state a preference to see the selected provider if there was more than one present, and if a GP asked why (since they were not known to them) the SP was trained to say, “I have a friend who saw you and said you were great.” In one event, the SP accidentally completed an interaction with a provider who was not consented and only realized the error after completing data entry. Since it is not possible to request consent after the fact, the team removed and deleted the data and undertook re-training of SPs to ensure that such an event would not happen again. This was effective when, for example, the study included two nearly-identical siblings with similar names and appearances at the same clinic, only one of who had given informed consent; the interaction was abandoned because the SP could not be sure of seeing the correct provider.
Discussion
South African law requires that freely given consent must always be at the forefront of research work. Regardless of the acceptability of SP work with a waiver of informed consent under U.S. regulation, ethical international collaboration requires that research activities be guided by respect for the population among which research is conducted. In South Africa, Section 12 of the constitution guarantees “freedom and security of the person,” stating explicitly that “Everyone has the right to bodily and psychological integrity, which includes the right [. . .] not to be subjected to medical or scientific experiments without their informed consent.” Ethical review boards as well as experts involved in research protocol development have repeatedly cited this basic right when refusing to support waivers of informed consent; Department of Health guidance is clear that this protection extends to all health research including service delivery. Hence, it is likely that a direct research study would be required to obtain informed consent from clinicians before observing them with SPs.
There is some room for secondary research conducted in collaboration with programs that are explicitly designed to ensure high quality medical care, such as in Christian et al. DOH guidance states that “[q]uality assurance and quality improvement studies (audits), programme evaluation activities and performance reviews usually do not constitute research and thus usually do not undergo formal ethics review. It should be noted, however, that if publication of such studies is desirable, it is prudent to obtain ethics approval before the study begins.” Afterward, it is possible that “the REC may approve a waiver of consent for secondary use of material or data where no more than minimal risk of harm is likely” (South Africa Department of Health, 2015). However, there may be additional hesitation when providing a waiver of consent to international study partners. Analyzing data based on the race of providers, for example, could fail to consider the fact that providers of color are more likely to practice in socio-economically disadvantaged communities, which means provider practice my differ based on the needs of their patients rather than competency levels.
For example, in Boffa et al., a qualitative component identified that providers who cater to cash-paying clients (an indication of socio-economic status) might first consider interventions that would save patients money. This included dispensing antibiotics before referring patients for a TB test, a practice discouraged by South Africa’s national TB program (Salomon et al., 2022). Some practitioners struggled to balance the consequences of delayed TB diagnosis with saving cash-paying clients the direct (private tests) or indirect (public referral) costs of excluding TB when the likelihood of an upper-respiratory tract infection seemed equally plausible. Ignorance of these realities creates a risk of research results and interpretations that are biased, judgmental, or simply incorrect.
It is clear from the experience of Boffa et al. that seeking explicit “opt-in” informed consent from providers to undertake SP work is time-consuming and costly; worse, it can preclude the ability of researchers to obtain accurate and actionable results for the public good. Over 40% of approached providers declined to participate, a proportion which makes representativeness of the sample very hard to support. It is not clear how these providers would have managed TB, making it difficult to draw conclusions about overall management practices, identify strengths or areas for improvement, or to compare to other settings. Similarly, when there is research interest in the performance of minority groups, excluding them from research may only continue to compound inequity and under-representation.
Others have suggested alternative approaches, such as “opt-out” consent in which a broad range of potential study subjects are informed of their potential selection for an SP study and given the opportunity to refuse participation through some official channel. We do not feel that this is likely to be appropriate outside of organized and trusted bureaucratic health care systems for a variety of reasons. Trust in strangers is often low; unsolicited, vague communication is likely to be ignored. In such a case, “a lack of refusal does not necessarily imply consent” (Rhodes and Miller, 2012). Furthermore, researchers might desire to include “traditional,” “informal,” “unofficial,” “illegal,” or other categories of health care providers outside “formal” medical establishments. There, such broad-stroke contacts from institutions such as universities or governments may cause active harm among communities of providers or care-seekers for a variety of reasons.
In order to maintain the scientific integrity of the SP method while remaining sensitive to contextual factors and potential for harm, we note here some alternative approaches that investigators may adopt in contexts where ethics boards may be reluctant to otherwise approve waivers of informed consent. First, it is critical to establish relevant research partnerships. Research teams should include representatives from all stakeholders and the research team must engage with groups who represent the population under study. In Boffa et al., the latter included liaising with independent practice associations focused on overcoming barriers to healthcare entry for doctors of color. As in Christian et al., it is also important to work closely with Departments of Health and local ethics bodies to ensure that the SP approach follows local requirements, that appropriate approvals are obtained, and that results and findings are produced for the benefit of the health system from which they were derived—not to advance researchers’ careers.
Honoring these rights and responsibilities would not be possible without the extensive local buy-in and consultation sought by a research team, and efforts to work with partner organizations that represented clinicians, especially in vulnerable groups. In South Africa, an independent practice organization that represents practitioners from disadvantaged populations was consulted during the research development process so that contextual factors were not overlooked and seemingly innocuous variables were not misrepresented. This was undertaken to ensure the participation and support of the community on design and methods, on outcomes of interest and interpretations (such as definitions of “correct management”), and in results dissemination.
Second, it is important to validate in every context, particularly when a study is in a novel setting or design. Boffa et al., like some other studies, included post-exit surveys and qualitative interviews with a sub-set of private providers to better understand the context under which participants practiced. Standardized patient studies that do not consider the context of practice may inadvertently interpret structural pressures or barriers as knowledge gaps, under-training, or incompetence—and risk drawing incorrect conclusions. Asking participants questions such as “why do you think some providers managed patients in this way?” can help uncover more accurate and representative explanations for management practices that are misaligned with practice guidelines. Some researchers have proposed going even farther—for example, by sharing with individual providers the exact details of their own performance. However, this has not yet been proposed or approved, as it is not clear how to properly protect that information, and all studies to date have upheld total anonymization and aggregation of published results.
Finally, findings should be reviewed by members of the participant population before presentation or publication. This could involve a presentation to a provider group before public dissemination, including a co-author from the provider group to weigh in on findings and their implications, or a workshop with study participants to consider alternate interpretations. Constituents—the people who form the participant population—should always benefit as a group from the knowledge generated by research. Adequate effort must be made to share the findings back to the research population before and after broader dissemination to ensure that they can incorporate lessons into their own practice. Thankfully, many of these practices are now gaining wide traction in international health research generally, and we encourage their extension and inclusion in SP studies (Morton et al., 2022).
Conclusion
Non-SP methods of collecting data about clinical practice exist, but they are often unable to answer the set of important research questions available in SP studies. Namely, non-audit observational methods cannot answer research questions that depend on knowing the likelihood that an individual with a certain condition will receive certain treatment by certain providers. Administrative data can only answer questions about how providers responded to certain presentations, but researchers cannot generally determine the individualized appropriateness of that care without invasively collecting protected health information from a large number of care-seekers to determine underlying need. Such an approach would likely pose even greater ethical and practical problems; we do not engage these here.
The simulation component of SP designs is one essential element to understanding why provider-centric approaches based on observed administrative practice data cannot provide a complete picture regarding the appropriateness of clinical management. While understanding clinical practice and patient presentation in general is undoubtedly important, such data cannot verify the appropriateness of management for a given individual care-seeker. With SP data, we know with certainty that the individual portrayed had a specific condition, and that they either did not receive specific types of care. We could, for example, argue that a provider’s practice was medically acceptable, or even exemplary, while simultaneously concluding that it resulted in incorrect care for a single patient (or vice versa). For example, SP data can determine how standardized diagnostic protocols for notifiable diseases such as tuberculosis affect costs or cause delays in diagnosis for patients who have other conditions such as asthma, COPD, or various other infectious conditions (King et al., 2021).
The complementary essential element to simulation is standardization. Non-SP patients are exceedingly unlikely to arrange their illnesses and choice of health care providers in ways that allow researchers to observe the relevant counterfactuals for the research question they are currently interested in, so generating the appropriate counterfactual situations is one of the core elements of SP research design. By repeating the carefully designed and simulated data-generative process using the same underlying scenarios across a specific set of providers of interest, or by varying elements of the scenarios, researchers can draw population-scale inferences about appropriateness of care, its variations, and its improvement that cannot be inferred from the distribution of care for non-SP patients. For instance, researchers have used SPs to investigate gender differences, variation by ethnicity, variation by patient demand, and variation by insurance status across multiple study sites (Currie et al., 2014; Daniels et al., 2019; Planas et al., 2015).
In the United States, it is well-established that IRBs are prepared to grant waivers of informed consent to SP studies that demonstrate minimal risk and programmatic need. Here, we have outlined empirical evidence that appropriately-designed SP studies can satisfy each of the five requirements of U.S. regulation for such a waiver. While there certainly are potential experimental manipulations of provider environments that would be possible, these go beyond the core data collection and must only be undertaken with informed consent. In addition, truly ethical research must go beyond a technical or legalistic interpretation of U.S. law, especially in international collaborations and post-colonial settings. Whether such waivers are appropriate (or whether they should be accompanied by additional conditions) will depend on each specific research context. Research teams must therefore carefully evaluate the ethical framework of non-U.S. study sites, engage deeply with communities where studies take place, and ensure that their work will result in benefits to those who participate in the research as well as the wider community.
Footnotes
Acknowledgements
The authors would like to acknowledge the contributions of our many collaborators, including but not limited to (alphabetically): Dr Jeremiah Chikovore, Dr Carmen Christian, Dr Amrita Daftary, Dr Jishnu Das, Ms Tsatsawani Mkhondo, Dr S Naidoo, Dr Madhukar Pai, and all our prior coauthors. We would like to thank the members of IRB and ethical review boards who have always provided insightful feedback and assistance. We further acknowledge the hard work, dedication, and insights of all the team members that have advanced this research, including all managerial and standardized patient staff.
Ethical approval
This study used only anonymized aggregated data from prior studies, therefore the authors declare that research ethics approval was not required for this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.