
Abstract
Background:
Vilaprisan is a selective progesterone receptor modulator with demonstrated efficacy in the management of uterine fibroids (UFs).
Objectives:
To evaluate the safety and efficacy of vilaprisan in Japanese women with UFs and heavy menstrual bleeding (HMB).
Design:
Open-label, parallel-group, Phase 3 randomized clinical trial.
Methods:
Japanese women with UFs and HMB were randomly assigned 1:1 to receive vilaprisan (2 mg/day) for either four treatment periods (TPs) of 12 weeks each separated by one bleeding period (Arm A1) or two TPs of 24 weeks each separated by two bleeding periods (Arm A2). The primary endpoint was the incidence of treatment-emergent adverse events (TEAEs).
Results:
Of 179 women enrolled, 151 were included in the full analysis set and 148 in the safety analysis set. TEAEs occurred in 79.1% of women, with the majority being mild; events were evenly distributed across both treatment arms. Study drug-related TEAEs were observed in 44.6% of women, and serious TEAEs were reported in 3.4% of women. During the treatment phase, the mean (standard deviation) number of bleeding days per 28 days decreased to 1.40 (1.34) days in Arm A1 and 1.42 (0.82) days in Arm A2 from respective baseline values of 5.1 (2.3) and 5.2 (2.0) days. Median time to onset of amenorrhea was 4 days in TP1 in both arms, and 4 days in TP2 in Arm A1. Absence of bleeding for the last 28 days was more common in TP1 (Arm A1: 91.89%, Arm A2: 89.19%) than TP2 (Arm A1: 80.85%, Arm A2: 85.71%).
Conclusion:
In this study, vilaprisan 2 mg/day was found to be well tolerated and efficacious in Japanese women with UFs and HMB. However, the study sponsor later terminated the overall clinical development of vilaprisan due to potential safety concerns from long-term rodent studies.
Registration:
The ASTEROID 8 study was registered at https://clinicaltrials.gov/ (registration number: NCT03476928).
Keywords
Introduction
Uterine fibroids (UFs) are common benign uterine smooth muscle tumors with a prevalence of approximately 70% among women, depending on race and age. 1 Approximately 25% of women with UFs develop symptoms that require treatment, most commonly heavy menstrual bleeding (HMB). 1 Women can also experience lower back pain and pelvic discomfort.2,3 reproductive issues, 2 and impaired quality of life (QoL). 3
Although surgical treatment of UFs is common, even minimally invasive interventions are associated with risks, and UF can recur requiring secondary interventions.4–7 Hysterectomy, a radical and invasive intervention for UFs, is associated with additional complications and is only suitable for women not wishing to preserve their fertility. 7
The Japan Society of Obstetrics and Gynecology and Japan Association of Obstetricians and Gynecologists 2017 guidelines recommend treatments such as estrogen-progestin combinations, gonadotropin-releasing hormone (GnRH) antagonists, tranexamic acid, and the use of a levonorgestrel-releasing intrauterine system for UF management. 8 However, GnRH antagonists are associated with hypoestrogenic side effects that necessitate the concurrent administration of hormonal add-back therapy. 9
Vilaprisan is an investigational selective progesterone receptor modulator (SPRM) with a potent anti-progestogenic effect 10 and demonstrated efficacy and tolerability in the management of UFs in Phase 1, 2, and 3 clinical trials.11–15 A 2-mg/day dose was selected for the Phase 3 studies in global and Japanese populations, based on exposure-response analysis. 16 The primary aim of the Phase 3 ASTEROID 8 study was to evaluate the safety of two treatment regimens of vilaprisan 2 mg/day in Japanese women with UFs and HMB.
The vilaprisan clinical program, including this Phase 3 trial, was paused in December 2018 to allow thorough evaluation of preliminary findings from long-term carcinogenicity studies in rodents that had been conducted, as standard practice, in parallel with the clinical program. Investigations revealed that these findings (increased occurrence of endometrial, adrenal, and skin tumors) were likely to be rodent-specific and of limited clinical relevance in humans. However, after discussion with the regulatory authorities, the study sponsor terminated the clinical program—including this study—after completion of an additional, comprehensive safety follow-up focusing on endometrial, adrenal, and skin safety (publications on these data are in development).
Materials and methods
Study design
ASTEROID 8 (ClinicalTrials.gov: NCT03476928) was a randomized, Phase 3, open-label, parallel-group, multicenter study conducted across 25 hospitals and clinics in Japan. Eligible women were randomly assigned 1:1 to Arm A1: Vilaprisan (2 mg/day) for four treatment periods (TPs) of 12 weeks (each separated by one bleeding period) or Arm A2: vilaprisan (2 mg/day) for two TPs of 24 weeks (separated by two bleeding periods); see Supplemental Figure 1).
The randomization sequence was generated by the sponsor’s Randomization Management group. Eligible subjects were enrolled by the study sites and were randomly and sequentially allocated within the strata via an interactive voice/web response system (IVRS/IWRS) in a 1:1 ratio to one of the two treatment groups, using block randomization with a block size of 2. Randomization was stratified by HMB during the screening period (>80.00 mL or not). At screening, upon signing the informed consent form and registering the patient in the IVRS/IWRS, each woman was assigned a unique multi-digit identification number by the site for unambiguous identification. Once allocated, this number was used to identify the woman throughout the study. Study sites assigned the correct treatment kit to each woman based on the randomization list. Participants and investigators were aware of the treatment, but central readers (bone mineral density (BMD) and endometrial biopsies) were blinded to treatment group and time point of assessment.
Sample size and early study closure
The sample size for ASTEROID 8 was based on the Pharmaceuticals and Medical Devices Agency regulatory requirements for submission to provide long-term safety data from at least 100 Japanese women with the target indication (i.e., UF and HMB) who were treated with the target regimen (i.e., A1 or A2) for at least 1 year, including treatment breaks. In this study, 75 women per regimen (150 women in total) were planned to be randomly assigned to the 2 treatment arms. Assuming a drop-out rate of 20%, 60 women per regimen (120 women in total) were planned to be treated for 1 year. In the ASTEROID 6 study (in submission), approximately 40 Japanese women, each with UF and HMB, were expected to be treated with the A1 and A2 regimen, respectively, for at least 1 year; thus, the overall planned number of Japanese women was approximately 100 per regimen (60 women from ASTEROID 8 plus 40 women from ASTEROID 6). If the number of enrolled Japanese women in ASTEROID 6 was higher than expected, recruitment in the ASTEROID 8 study was planned to be stopped before reaching the overall target of 150 randomly assigned women, because 100 women per regimen from the two studies would be sufficient to fulfill the regulatory requirement for long-term safety evaluation in Japanese women. Conversely, in the event of lower enrollment in ASTEROID 6, the number of women enrolled in ASTEROID 8 would be increased. However, due to the premature closure of the vilaprisan clinical trial program (details below), only 153 women overall out of 200 planned were randomly assigned.
The trial began in 2018 with the first woman enrolled on 30 March; the last woman’s last visit occurred in July 2021. Recruitment and the start of new TPs were paused in December 2018 due to pre-clinical findings from 2-year rodent carcinogenicity studies showing adrenal, uterine, and skin abnormalities after lifelong vilaprisan exposure. At the time of the study pause, women who were already randomly assigned but had not yet entered TP1 were instructed to not start treatment, whereas those already taking the study treatment were instructed to complete their current TP but to not start a new one. At that time, the aim was to restart the study once the pre-clinical results were thoroughly evaluated. Investigations identified that the results were likely to be rodent-specific and, therefore, of limited clinical relevance in humans. However, the study sponsor later elected to close the study after these preclinical investigations, as discussions with regulatory authorities had resulted in a prolonged treatment break in the clinical trial. To ensure the safety of enrolled women and thoroughly assess the potential relevance of the preclinical findings to humans, all women who had received at least one dose of vilaprisan were asked to participate in a comprehensive safety follow-up as part of a safety closeout visit (SCOV), focusing on endometrial, adrenal, and skin safety. Results from safety evaluations were later communicated to women during a safety result reporting visit (comprehensive publications on these safety data are in development).
The collection of efficacy and safety data for the TP was not impacted by the COVID-19 pandemic as the trial was already on a temporary pause by then. At this time, women were in the safety follow-up period. However, the COVID-19 pandemic caused delays in the SCOV, and onsite monitoring after study closure was confirmed.
Ethics approval and consent to participate
The reporting of this study conforms to the CONSORT 2025 statement for randomized clinical trials. 17 The study was approved by each of the study center’s Institutional Review Board (IRB). Appendix I in the Supplemental Material provides a list of the IRBs for the ASTEROID 8 study (approval numbers are documented in the local trial files and are available on request). The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonization Guideline E6: Good Clinical Practice. Prior to enrollment in the study, all participants provided written informed consent to conduct the study and publish anonymized data related to the study.
Study population
ASTEROID 8 enrolled adults with UF(s) documented by ultrasound at screening. Participants were required to have HMB (defined as menstrual blood loss (MBL) > 80.00 mL measured by the menstrual pictogram (MP) method) in a bleeding period during screening or self-reported HMB during three bleeding episodes within 6 months prior to screening. Women were also required to have good general health (except for UFs) and otherwise normal endometrial histology based on a biopsy performed at screening. All women were required to use a non-hormonal contraceptive method. Key exclusion criteria included pregnancy or lactation, undiagnosed abnormal genital bleeding, and concomitant treatment(s) that could have interfered with the conduct of the study or interpretation of results (e.g., hormonal contraceptives, tranexamic acid, GnRH antagonists, or anticoagulants).
Study endpoints
The primary endpoint was the incidence of treatment-emergent adverse events (TEAEs), and the secondary endpoint was the mean number of bleeding days normalized by 28 days. Other endpoints included HMB response (MBL of <80.00 mL during the last 28 days of TP1 and >50% reduction versus baseline), time to onset of amenorrhea (time until the first day for which MBL for all subsequent 28-day periods up to the end of the TP was <2 mL), time to onset of controlled bleeding (time until the first day for which MBL for all subsequent 28-day periods up to the end of the TP was <80 mL), and absence of bleeding (no bleeding (spotting allowed) during the last 28 days of the TP).
Safety monitoring
Safety parameters including adverse events (AEs), TEAEs, and laboratory evaluations were regularly and closely monitored throughout the study. Incidences of TEAEs, AEs, drug-related TEAEs, and serious AEs (SAEs) were summarized using frequency tables based on the Medical Dictionary for Regulatory Activities (MedDRA version 24.0) preferred terms (PTs) within the primary System Organ Class. TEAEs were defined as any AEs that occurred after the first study drug intake until 60 days after the last intake; AEs that started after 60 days were defined as post-treatment AEs. TEAEs that occurred during the TP until the date of the last study drug intake plus 8 days were considered “on-treatment” (based on the half-life of vilaprisan), and all other TEAEs were listed as “off-treatment.” AEs of special interest were categorized and reported based on MedDRA groupings using MedDRA version 24.0.
Endometrial safety included regular ultrasound investigations during the TPs (Arm A1: Screening, randomization, Week 13 of TP1, Week 13 of TP2, and at the SCOV; Arm A2: Screening, randomization, Week 13 of TP1, Weeks 4–5 after TP1, Week 13 of TP2, and at the SCOV), observation of bleeding patterns, and endometrial biopsies (at screening and the SCOV), which were assessed centrally by three independent expert pathologists. Unscheduled biopsies were performed in women with increased endometrial thickness of >18 mm or a suspicious bleeding pattern, for example, continuous spotting or unusually heavy bleeding after the start of treatment. Monthly monitoring of liver parameters was implemented for the initial year of treatment, in accordance with the Food and Drug Administration 2009 drug-induced liver injury guideline. 18 A liver symptom questionnaire was introduced early in the study, based on a liver-related safety signal observed with another SPRM, ulipristal acetate. 19
BMD (spine, hip, and femoral neck) was assessed using dual-energy X-ray absorptiometry (DEXA) at screening and during the “BMD visit” that occurred at Weeks 50–54 after the start of TP1. At each DEXA scan, two measurements were performed for each location using the same device, and the mean values were used for evaluation. Women who received vilaprisan treatment also had an off-treatment DEXA scan performed at the SCOV to confirm absence of bone loss, or recovery therefrom.
After the decision to close the study, additional endometrial, skin, and adrenal monitoring was introduced as part of the comprehensive safety follow-up. Detailed descriptions of these assessments and findings from these safety analyses across the vilaprisan clinical trial program will be published separately.
Efficacy assessments
Women were required to complete an electronic diary (eDiary) daily throughout the study. Controlled bleeding and absence of bleeding were assessed using data collected from the Uterine Fibroid Daily Bleeding Diary as part of the eDiary, whereby women documented the severity of their vaginal bleeding each day (no bleeding, spotting, mild, moderate, severe, or very severe). Women also documented their MBL per sanitary product using the MP visual scoring system in the eDiary, and these data were used to assess HMB response and amenorrhea rate.
Statistical analyses
No statistical hypotheses were planned to be tested; all analyses were descriptive. All safety analyses were performed on the safety analysis set (SAF), and BMD analyses additionally on the modified SAF. The SAF comprised all women administered with at least one dose of vilaprisan; the modified SAF comprised all women in the SAF without any validity findings that may have potentially affected BMD. Validity findings leading to exclusion from the modified SAF were specified in the “Specification of assessment criteria and identification requirements,” which was finalized before the database lock. The full analysis set (FAS) was used for the analysis of all other variables and comprised all randomly assigned women who started TP1 (including those who did not take the study drug). Time-to-event variables were analyzed using Kaplan–Meier estimates. Statistical analysis was performed using the software package, SAS release 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient disposition
The ASTEROID 8 clinical trial took place between March 2018 and July 2021. Overall, 179 women were enrolled from 25 study centers in Japan. Of these women, 153 (85.5%) were randomly assigned to treatment (76 to Arm A1; 77 to Arm A2). The most common reason for women not being randomly assigned was screening failure (n = 21, 11.7%). Overall, 151 (98.7%) women were included in the FAS, 148 (96.7%) were included in the SAF (n = 74 per arm), and 147 in the modified SAF (73 in Arm A1; 74 in Arm A2); 47 and 70 women in Arm A1 and Arm A2, respectively, completed TP2. For more details on patient disposition, see Figure 1.

Patient disposition.
Demographics and baseline characteristics
The women’s baseline demographics and disease characteristics in the FAS were generally comparable between the two treatment arms, with an overall mean (standard deviation (SD)) age of 42.3 (5.2) years, MBL volume of 159.8 (104.0) mL, and the largest UF diameter of 42.0 (23.6) mm (Table 1). The mean (SD) volume of the three largest UFs was numerically higher in the A1 arm versus the A2 arm (148.7 (236.4) mL versus 127.3 (208.3) mL, respectively).
Demographics and baseline characteristics (full analysis set).

MP: menstrual pictogram; SD: standard deviation.
Safety analyses
Overall, AEs were reported in 143 (96.6%) of the 148 women in the SAF: n = 71 (95.9%) in Arm A1 and n = 72 (97.3%) in Arm A2 (see Table 2).
Summary of AEs and TEAEs (safety analysis set).
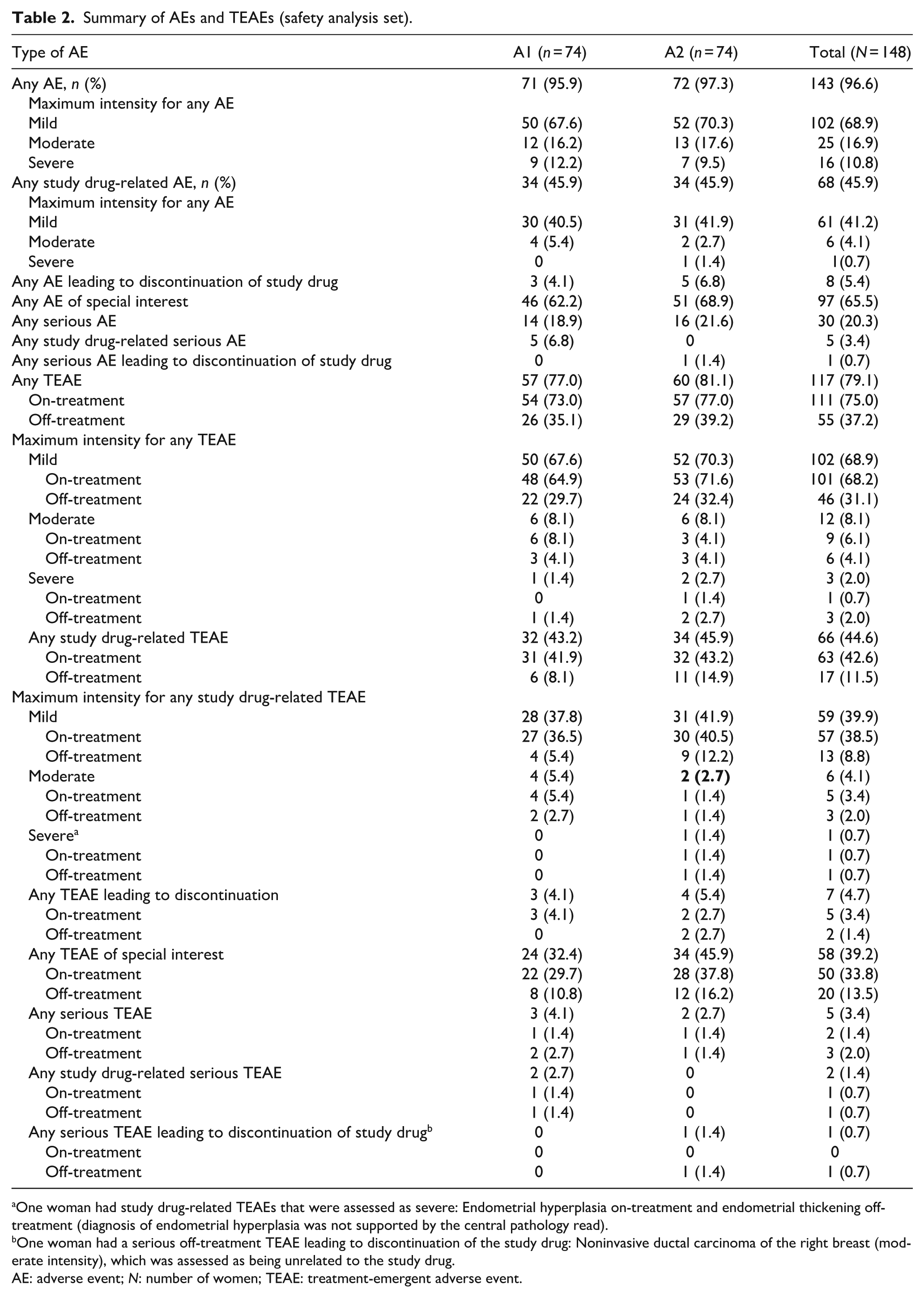
One woman had study drug-related TEAEs that were assessed as severe: Endometrial hyperplasia on-treatment and endometrial thickening off-treatment (diagnosis of endometrial hyperplasia was not supported by the central pathology read).
One woman had a serious off-treatment TEAE leading to discontinuation of the study drug: Noninvasive ductal carcinoma of the right breast (moderate intensity), which was assessed as being unrelated to the study drug.
AE: adverse event; N: number of women; TEAE: treatment-emergent adverse event.
Primary safety endpoint: Incidence of TEAEs
A summary of the AEs and TEAEs is shown in Table 2. TEAEs were reported for 117/148 (79.1%) women: n = 57 (77.0%) in Arm A1 and n = 60 (81.1%) in Arm A2. The majority of TEAEs were mild in intensity (68.9%). Overall, the most common TEAEs were nasopharyngitis (n = 45 (30.4%)), intermenstrual bleeding (n = 29 (19.6%)), hot flush (n = 20 (13.5%)), and headache (n = 14 (9.5%)); see Supplemental Table 1.
Study drug-related TEAEs were reported for 66 (44.6%) women: n = 32 (43.2%) in arm A1 and n = 34 (45.9%) in arm A2; Table 2. On-treatment study drug-related TEAEs occurred in 63 (42.6%) women, and off-treatment study drug-related TEAEs were reported in 17 (11.5%) women. The majority of study drug-related TEAEs were mild in intensity (39.9%). Overall, the most common study drug-related TEAEs were intermenstrual bleeding (n = 28 (18.9%)) and hot flush (n = 19 (12.8%); Supplemental Table 2).
TEAEs leading to discontinuation of the study drug occurred in seven women (n = 3 (4.1%) in Arm A1; n = 4 (5.4%) in Arm A2); these TEAEs occurred on-treatment in five (3.4%) women and off-treatment in two (1.4%) women (Table 2 and Supplemental Table 3).
Five (3.4%) women reported serious TEAEs: three (4.1%) women in Arm A1 and two (2.7%) women in Arm A2 (Table 2 and Supplemental Table 4). Serious on-treatment TEAEs occurred in two (1.4%) women and serious off-treatment TEAEs occurred in three (2.0%) women. Serious TEAEs were assessed as study drug-related (hypopituitarism (off-treatment) and peritonitis (on-treatment)) for two (1.4%) women in Arm A1. One woman in Arm A2 had TEAEs that were assessed by the study investigator as severe and study-drug related: endometrial hyperplasia on-treatment and endometrial thickening off-treatment (the diagnosis of endometrial hyperplasia was not supported by the central pathology read); neither TEAE was considered serious, and both were later reported to be resolved. One serious off-treatment TEAE led to the discontinuation of the study drug in Arm A2; the woman was found to have non-invasive ductal carcinoma of the right breast (moderate intensity), which was classified as serious due to hospitalization, and the event was later assessed as being unrelated to the study drug by the investigator and study sponsor. No deaths were reported in the study.
Overall, post-treatment SAEs (starting 61 days after the last study drug intake or later) were reported for 26 (17.6%) women (n = 11 (14.9%) in Arm A1; n = 15 (20.3%) in Arms A2; Supplemental Table 5). The most frequent PT was myomectomy for eight (5.4%) women (in 4 (5.4%) women per treatment arm). Three (2.0%) post-treatment SAEs were assessed as study drug-related in Arm A1 (blood aldosterone increased in one (0.7%) woman and adrenal adenoma in two (1.4%) women).
Secondary efficacy endpoint: Number of bleeding days
The mean (SD) number of bleeding days per 28 days was 1.40 (1.34) days in Arm A1 and 1.42 (0.82) days in Arm A2, decreasing from respective baseline values of 5.1 (2.3) and 5.2 (2.0) days, respectively (Figure 2).

Number of bleeding days at baseline (days 1–28) and by UF-DBD normalized by 28 days.
Other efficacy endpoint: HMB response
In Arm A1, HMB response was reported more frequently in TP1 (n = 72/74 (97.30%)) than in TP2 (n = 39/47 (82.98%)). In Arm A2, HMB response was similar in TP1 (n = 68/74 (91.89%)) and TP2 (n = 64/70 (91.43%)).
Other efficacy endpoint: Time to onset of amenorrhea
The median time to onset of amenorrhea for TP1 was 4 days in both arms. The overall median in TP2 was 4 days in Arm A1 (not applicable for Arm A2).
Other efficacy endpoints: Time to onset of controlled bleeding and absence of bleeding
The median time to onset of controlled bleeding for TP1 was 1 day for both arms. The overall median in TP2 was 1 day in Arm A1 (not applicable for Arm A2).
Absence of bleeding (spotting allowed) during the last 28 days of treatment in TP1 was reported in 68 (91.89%) women in Arm A1 compared with 66 (89.19%) women in Arm A2. In TP2, the proportion of women with no bleeding (spotting allowed) per arm were 80.85% in Arm A1 and 85.71% in Arm A2.
Safety: Endometrial thickness, liver monitoring, estradiol, and BMD
Mean endometrial thickness in both treatment arms decreased from baseline during treatment and returned to near baseline values during follow-up (Supplemental Table 6). No combined elevated hepatic safety parameters were reported throughout the study, and there were no cases that met Hy’s law criteria. 20
Mean (SD) baseline estradiol values for arms A1 and A2 were 144.88 (115.44) and 161.94 (115.39) pg/mL, respectively. Values decreased to 102.13 (93.00) pg/mL during the first bleeding break (Days 1–28) in Arm A1 and 63.83 (68.01) pg/mL during TP2 (Days 1–28) in Arm A2. During follow-up (Days 57–84), values increased to 205.87 (353.14) and 129.41 (96.71) pg/mL, respectively. After this, a trend toward a return to baseline levels was observed in both Arms.
BMD changes from baseline for the modified SAF (women with available data in the electronic case report form, n = 147) are reported in Supplemental Table 7. However, it is noted that interpretation of these data at the SCOV should be conducted with caution due to the delayed timepoint of measurement that resulted from the premature discontinuation of the study.
Safety: Assessments at the SCOV
Of the 119 women who consented to the SCOV (n = 57 in Arm A1; n = 62 in Arm A2), two (1.7%) women (both in Arm A1) were diagnosed with non-functional benign cortical adenoma. Other adrenal disorders were specified as subclinical Cushing’s syndrome in two women (one woman per arm).
No cases of malignant adrenal tumors were reported, and no signal for adrenal safety was identified from the study. No cases of malignant uterine neoplasm or endometrial hyperplasia with or without atypia were reported in the central pathology reads, and no pre-cancerous skin lesion, cutaneous sarcoma, or any other malignant skin tumor was reported in this study. Furthermore, there were no women with an endometrial thickness >18 mm at the SCOV.
Discussion
In the ASTEROID 8 study, a comprehensive safety monitoring program demonstrated that two treatment regimens of vilaprisan 2 mg in Japanese women were well-tolerated. There were no unexpected trends observed in the reported TEAEs (primary safety variable) in either treatment arm, or for drug-related AEs and other SAEs, and the events were similar to those observed in other Phase 3 studies of vilaprisan.11,12,15
TEAEs were evenly distributed across the two treatment arms, and most women had mild TEAEs and mild study drug-related TEAEs; only one woman had a study drug-related TEAE that was assessed as being severe. Although the rate of TEAEs was high in both treatment arms, this was consistent with findings from previous studies of SPRMs and GnRH antagonists for UF management9,11,12,15,21–24; the proportions of women with severe TEAEs, serious TEAEs, and study drug-related serious TEAEs were notably lower. A small number of women discontinued the study drug due to TEAEs, for which a similar number of on-treatment and off-treatment SAEs were reported. Overall, the most common TEAEs were nasopharyngitis, intermenstrual bleeding, hot flush, and headache, which is comparable with findings in previous studies. The most frequently reported post-treatment PT was myomectomy, reflecting a feature of the underlying disease after the study treatment was ceased.
Based on the United States’ Food and Drug Administration 2009 drug-induced liver injury guideline 18 and reports of a serious liver-related safety signal observed with another SPRM, ulipristal acetate, 19 a liver symptom questionnaire and monthly monitoring of liver parameters were implemented in ASTEROID 8. No clinically relevant changes were detected in liver function, which was consistent with safety outcomes reported in previous vilaprisan studies.11–15 For ulipristal acetate, data from the PEARL clinical trials did not indicate concerns regarding potential liver injury; however, there were several postauthorization reports of severe liver injury worldwide, which necessitated a liver transplant in four women and subsequently led to the withdrawal of ulipristal acetate in Canada, limitation of its use in Europe (to premenopausal women ineligible for surgery or with unsuccessful surgical outcomes), and lack of consideration for approved in the United States for UF treatment.6,19,25,26 Based on the clinical trial findings, it was concluded that although ulipristal acetate does not belong to a class of medications known to cause drug-induced liver damage, some patients may develop idiosyncratic drug-induced liver damage for which there are no known identifying markers available. 19 Thus, further data from larger populations may be needed to confirm the liver safety profile of vilaprisan.
The vilaprisan clinical program, including this Phase 3 trial, was paused in 2018 based on preliminary findings from long-term rodent carcinogenicity studies and was later terminated. These findings in rodents included the increased occurrence of endometrial, adrenal, and skin tumors. In ASTEROID 8, endometrial monitoring confirmed no cases of malignant neoplasm or endometrial hyperplasia with or without atypia in the central pathology reads, and results derived from adrenal monitoring were in line with the expected background rate of incidentally discovered adrenal masses in the study population. 27 In addition, no malignant skin tumors or precancerous skin lesions were reported.
The secondary efficacy variable, mean number of bleeding days per 28 days, showed substantial reductions versus baseline, and this result was very similar between the two treatment arms. Furthermore, a high HMB response rate, amenorrhea rate, and absence of bleeding rate were reported in both treatment arms, with no marked differences observed.
ASTEROID 8 enrolled a broad population of women in respect to volume of MBL and fibroid size, thereby providing valuable insights into a representative, real-life population of Japanese women with UFs. Furthermore, more than 55% of the women fulfilled the inclusion criteria for the population defined for other ASTEROID efficacy studies, which allowed a meaningful efficacy analysis. There were no differences in baseline characteristics and disease demographics between the treatment arms.
A 2015 report estimated that approximately 550,000 women have received treatment for UFs in Japan. 28 The true prevalence of UF is likely to be higher because, despite experiencing severe menstrual symptoms, Japanese women often refrain from consulting gynecologists. Encouraging women to seek help can improve healthcare access and outcomes and improve QoL.28,29 In addition, East Asian women have a higher disease burden associated with UFs compared with White women. 30 These variations highlight the importance of obtaining accurate disease burden data, which can assist policymakers and governments in tailoring healthcare strategies for specific populations. 31
Study limitations
The initially planned 1-year TP was not completed due to the early discontinuation of the study; the average treatment duration was instead approximately 6 months, and this shortened exposure duration would have reduced the likelihood of detecting rare or late-onset AEs. Despite the shorter overall study duration, the initially planned enrollment target was achieved, and most women consented to the SCOV. The additional monitoring implemented in the SCOV of ASTEROID 8 was based on guidance from regulatory authorities and provided further reassurance that the observed preclinical data do not translate into the human setting.
Further limitations of the study are that the study was open-label, no comparator arm was included, and the sample size was determined based on regulatory requirements for long-term safety data instead of using a formal sample size calculation; therefore, the findings reported here need to be interpreted cautiously and within the context of previous studies.
Conclusion
Results from the ASTEROID 8 study indicate that vilaprisan administered at 2 mg/day for TPs of 12 or 24 weeks is well tolerated in Japanese women, and these findings are consistent with those of other vilaprisan clinical trials that enrolled global populations of women.11,15,21,24 Despite the early termination of the development program for vilaprisan, this study provides promising safety and efficacy findings to support and encourage the continued development of SPRMs for the treatment of women with UFs.
Supplemental Material
sj-docx-1-whe-10.1177_17455057251378954 – Supplemental material for Safety and efficacy of vilaprisan in Japanese women with fibroids: The Phase 3 ASTEROID 8 trial
Supplemental material, sj-docx-1-whe-10.1177_17455057251378954 for Safety and efficacy of vilaprisan in Japanese women with fibroids: The Phase 3 ASTEROID 8 trial by Mikio Momoeda, Thomas Faustmann, Esther Groettrup-Wolfers, Masami Kondo, Masanobu Yasuda and Christian Seitz in Women's Health
Footnotes
Acknowledgements
The authors thank study investigators, coordinators, and nurses, and the women for their contributions. Dr Kelly Genga, MD, PhD, assisted with review of the safety data in the article. Medical writing and editorial support for the preparation of this article, under the direction of the authors, was provided by Sinéad Mutton, MSc, of ApotheCom, an Inizio Company, and was funded by Bayer AG.
Ethical considerations
The study was approved by each of the study center’s Institutional Review Boards (IRBs). ![]() provides a list of the IRBs for the ASTEROID 8 study (approval numbers are documented in the local trial files and are available on request). The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonization Guideline E6: Good Clinical Practice.
provides a list of the IRBs for the ASTEROID 8 study (approval numbers are documented in the local trial files and are available on request). The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonization Guideline E6: Good Clinical Practice.
Consent to participate
All women provided written informed consent to participate in the study.
Consent for publication
All women provided written informed consent to publish anonymized data related to the study.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Bayer AG, Berlin, Germany, including the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the article; and decision to submit the article for publication. Medical writing support was provided by ApotheCom, an Inizio Company, with funding from Bayer AG, in accordance with Good Publication Practice guidelines.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.M. has no competing interests to disclose; E.G.-W. and C.S. are full-time employees of Bayer AG., Berlin, Germany; T.F. is a former employee of Bayer AG; M.K. and M.Y. are full-time employees of Bayer Yakuhin Ltd., Osaka, Japan.
Data availability statement
Availability of the data underlying this publication will be determined according to Bayer’s commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing.” This pertains to scope, timepoint, and process of data access. As such, Bayer commits to sharing, upon request from qualified scientific and medical researchers, patient-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in women for medicines and indications approved in the United States (US) and European Union (EU), as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 1, 2014. Interested researchers can use ![]() to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the member section of the portal. Data access will be granted to anonymized patient-level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the member section of the portal. Data access will be granted to anonymized patient-level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.