
Abstract
Peripheral nerve injury activates microglia in the spinal, promoting microglial polarization and facilitating neuropathic pain progression. Necroptosis, a form of cell death, plays a crucial role in various neurological diseases and receptor-interacting protein kinases 3(RIPK3) a key molecular in the process. This study investigates to explore that RIPK3 regulates microglial polarization through the TLR4/MyD88 signaling pathway in neuropathic pain. By using a chronic constriction injury (CCI) model in mice, we found that peripheral nerve injury promoted M1 polarization and activated the TLR4/MyD88 pathway in spinal cord; in BV-2 microglia models, TNF-α/Z-VAD co-induction triggered M1 polarization through TLR4/MyD88 pathway, TLR4 antagonists suppressed these effects both in vivo and in vitro. Administration of GSK’872 (RIPK3 inhibitor) inhibited TLR4/MyD88 pathway, reduced microglial M1 polarization, promoted microglial M2 polarization and alleviated CCI-induced hyperalgesia. These findings suggest that necroptosis is a key cellular mechanism in peripheral injury-induced neuropathic pain and that RIPK3 regulates microglial polarization via the TLR4/MyD88 pathway, providing a new target for neuropathic pain treatment and clinical prevention.
Introduction
Neuropathic pain is a chronic disease that seriously impacts people’s life. It causes profound physiological and psychological harm, functional impairment, and increased complications. 1 According to surveys, neuropathic pain affects 480–560 million people worldwide, and its prevalence in the general population is estimated to be 7%–10%, 2 but current therapies often fail to provide adequate relief, elucidating its underlying pathophysiological mechanisms is essential for developing effective treatments.
The necroptosis pathway, primarily involves receptor-interacting protein kinase 1(RIPK1)- receptor-interacting protein kinase 3 (RIPK3) -mixed lineage kinase domain-like protein (MLKL).3,4 This pathway is not only associated with the onset and progression of multiple diseases, such as neurodegenerative diseases and ischemia-reperfusion injury,5,6 but it often leading to an inflammatory response.7,8 In neuropathic pain, this process contributes to neuroinflammation and exacerbates pain hypersensitivity. The RIPK3 kinase plays a pivotal role in mediating necroptosis, RIPK3 triggers a signaling cascade that results in the phosphorylation of MLKL, which then disrupts cellular membranes and causes necroptosis. 9 This release of intracellular contents acts as damage-associated molecular patterns (DAMPs), further activating immune responses and sustaining inflammation.
Neuropathic pain is a universal, severe, chronic disease, with pathogenesis centered on the central nervous system. Many immune cells are involved in developing neuropathic pain caused by nerve injury. 10 Neuroimmune activation has been identified as a process that plays an important role in neuropathic pain. 11 By establishing a rodent model of neuropathic pain, the relationship between microglia activation and pain behavior was confirmed. 12 Microglia are very sensitive to changes in the extracellular environment, and when the body is subjected to ischemia, trauma, and infections, microglia transform from a resting state to an activate state. Following a model of peripheral injury, microglia in the dorsal horn of the spinal responded very rapidly and underwent marked changes in morphology and number that are necessary for the development of pain. 13 Typically, M1-type microglia, activated by exogenous stimuli, can enhance neuronal excitability, leading to nociceptive hypersensitivity and promoting neuropathic pain. In the chronic phase, the persistent activation of M1-type microglia leads to the prolonged presence of neuroinflammation, which in turn prolongs and exacerbates the pain response. This persistent inflammatory environment exacerbates neuronal excitability and leads to refractory pain. 14 In contrast, M2-type microglia secrete anti-inflammatory factors that inhibit M1 responses and reduce neuronal excitability and pain. Experimental studies have shown that promoting M2 polarization can reduce neuropathic pain.15,16 Therefore, microglia play a very important role in the development of neuropathic pain.
Toll-like receptor 4 (TLR4) is a critical transmembrane protein molecule for signal transduction, and Myeloid Differentiation Primary Response Protein 88 (MyD88) is a downstream gene of each subtype of TLRs, which can participate in the body’s intrinsic immunity through the regulation of nuclear factor (NF-kB) and other pathways. Previous experiments have shown that TLR4 expression is highly increased in the spinal cord of mice during CCI injuries.17,18 The TLR4/MyD88 has role in regulating M1/M2 polarization. 19 TLR4 activates NF-kB transcription via MyD88, releasing pro-inflammatory factors that drive microglia to M1-type polarization, exacerbating neurological damage.20,21 In certain pathological conditions, such as Alzheimer’s and Parkinson’s disease, over-activation of M1-polarized microglia exacerbates neuronal death and disease progression. Blocking TLR4 or downregulating MyD88 signaling attenuates the M1-type response, converting microglia to the M2-type and enhancing their repair function. For example, certain natural compounds and synthetic small molecule inhibitors have demonstrated the potential of inhibiting TLR4/MyD88 signaling to reduce both neuroinflammation and neuropathic pain. 22 Microglial polarization can cause neuroinflammation and retinopathy through activation of the TLR4/MyD88 pathway. 17
In this study, we aimed to explore the role of necroptosis in neuropathic pain using mice peripheral nerve injury model. We researched the molecular mechanisms by which RIPK3 regulates the M1/M2-polarization state of microglia through the TLR4/MyD88 signaling pathway to provide a new theoretical basis for the neuropathic pain pathogenesis to offer a new theoretical basis (Figure 1).
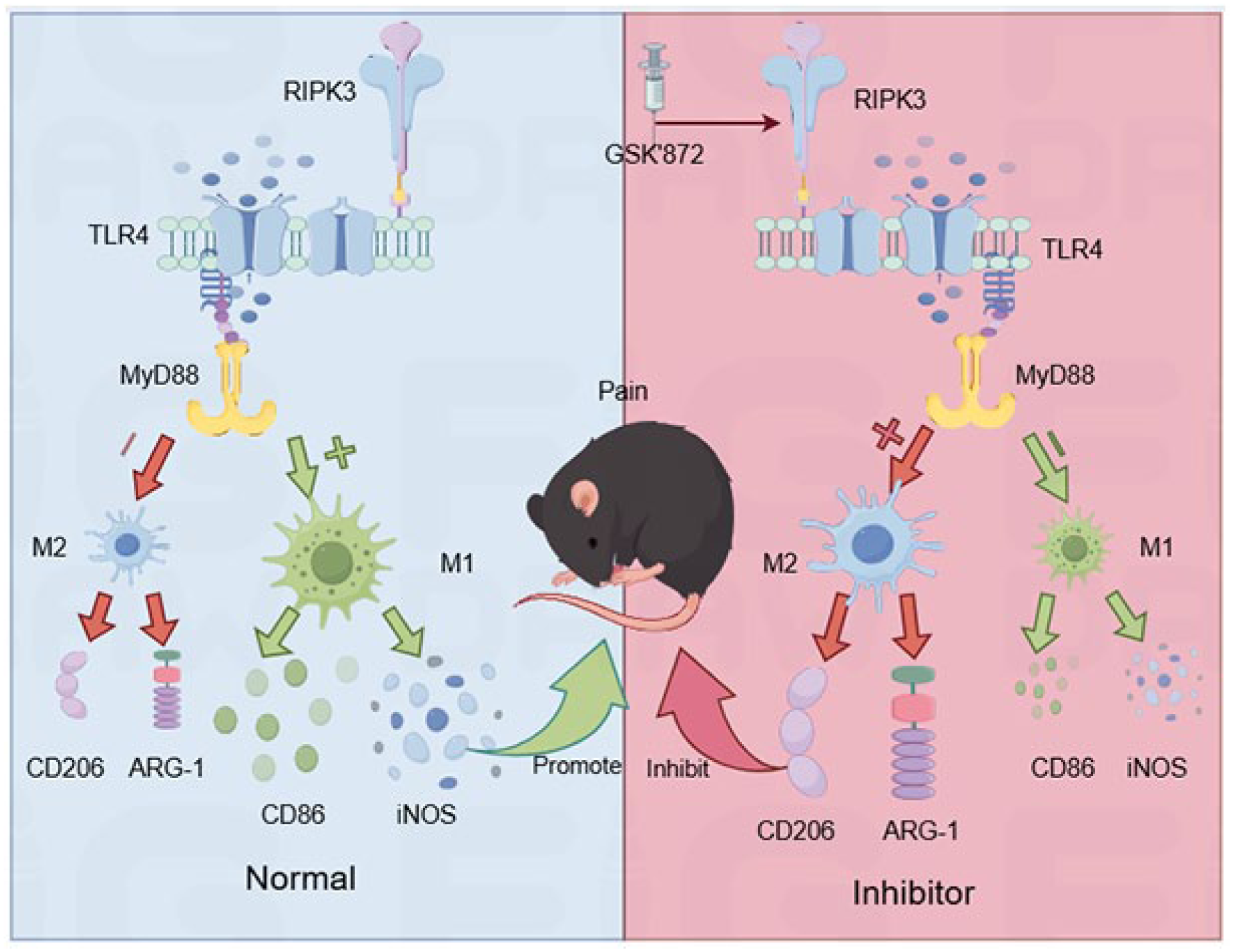
Mechanism of RIPK3/TLR4/MyD88 pathway in microglial polarization in neuropathic pain.
Materials and methods
Animals
Adult male C57BL/6J mice (9–12 weeks) were obtained from the Laboratory Animal Center of Shandong Second Medical University. The mice were kept in a constant temperature and humidity environment with light and dark cycles for 12 h. Animal Research Guidelines conducted the study: Reporting of in Vivo Experiment (ARRIVE). The Ethics Committee of Shandong Second Medical University approved the animal experiments.
Treatments and experimental protocols
All mice were anesthetized via 1% sodium pentobarbital (60 mg/kg) via intraperitoneal injection. In brief, the right sciatic nerve was separated at the midthigh level and loosely ligated at 1 mm intervals using four chromic catgut sutures. It was ensured that the hind limb briefly twitched, and the nerve was not squeezed too tightly to prevent blood flow around the nerve. The muscles and skin were sutured in layers. 23 In Sham group, the right sciatic nerve was exposed but not ligated. The surgical mice were randomly assigned to one of the following groups (n = 10): Sham, CCI, CCI + DMSO, and CCI + 1 mg/kg GSK 872 (25 mg, Sigma, USA). 24 Dimethyl sulfoxide was used to dissolve GSK’872. Equal volumes of DMSO were administered to mice in the CCI+DMSO group in a similar manner. After behavioral tests, the animals used for the study were euthanized using an overdose of anesthetic. Subsequently, we collected spinal cord tissue and stored it at −80℃ for future use.
Behavioral tests
Assessment of mechanical allodynia
Four groups of mice were weighed 1 day before the experiment and 1, 3, 7, 10, and 14 days after the operation, and then the wire mesh was placed at a certain height A 20-cubic-centimeter square mice box was prevented from the wire mesh, which was sealed and invisible all around, with holes on the top to be transparent, and the mice were put into the box to adapt to the mice box for 30 min before each experiment. Starting from 0.4 g, we vertically stimulated the middle skin of the plantar surface of the right hind limb, and mice were stimulated in the same position so that the fiber wire was bent into a “C” shape and held for 5 s. If the mice did not have a paw-shrinking response, we switched to the next strength, that is, 0.6 g fiber wire, to stimulate the hind paw; and at this time, if the mice had a paw-shrinking response, then we would select the following strength, that is, 0.16 g. To stimulate the hind paw of the mice. According to this method, when a response different from the previous one appears, continue to promote four times according to the above process, plus the first two times, a total of six effective stimulations, mice with the same foot for each stimulation should be separated by 30 s. The calculation was done by adopting the up-and-down method.
Assessment of thermal hyperalgesia
Similar to the Von Frey experiment, the same square mice box was also placed on the glass shift of the radio thermal analgesia meter, and the experimental mice were first put in to acclimatize for 30 min and then irradiated with the thermal radiometer to the middle of the hind foot of the mice, and the time from the beginning of irradiation to the appearance of paw-shrinking response, that is, the leg-shrinking latency period, was recorded. The test was repeated, and the light automatic stopping time was set to 20 s to prevent the mice from being scalded on the soles of their feet. Mice were repeated five times, with each stimulation interval of 5 min. The maximum and minimum values were removed from the obtained five test times, and the average value of the three times was taken as the thermal pain threshold of the mice.
Tissue preparation and immunofluorescence
Mice were anesthetized by injection and the heart exposed, then treated by perfusion of pre-cooled saline and paraformaldehyde, and eventually, the L4–L6 segments of the spinal cord were removed and placed in 4% paraformaldehyde for fixation and disposal, followed by 30% sucrose dehydration and incubation at 4℃ for dehydration until the tissue was deposited at the bottom of the centrifuge tube. The spinal cord tissue was placed vertically on a specimen tray, an OCT embedding agent was added and waited for it to freeze and harden completely, and the embedded tissue was cut crosswise to a thickness of 12 μm and pasted onto an adhesive slide. After washing with PBS three times, each time for 5 min, it was osmotically treated with 0.3% TritonX-100 for 15 min and closed with 10% normal serum at room temperature for 1 h. Excess serum was removed, and appropriate ratios of primary antibodies were added dropwise, rabbit anti-RIPK3 (1: 200 dilution, ab255705, Abcam, USA) with mice anti-Iba-1 (1: 400 dilution, ab178846, Abcam, USA) or mice anti-GFAP (1:200 dilution, AF0156, Reyotime, China) or mice anti-NeuN (1:200 dilution, 66836-1-Ig 66836-1-Ig, Proteintech, USA), incubated overnight at 4℃; primary antibody was recovered, washed three times with PBS for 5 min, and then mixed secondary antibody of CoraLite488-labeled goat anti-rabbit IgG (1:100 dilution, 10209-2-AP, Proteintech, USA) and CoraLite594-labeled goat anti-mice IgG (1:500 dilution, AB_2810984, Proteintech, USA) was added dropwise, incubated for 30 min at 37℃, protected from light at room temperature; secondary antibody was recovered and washed three times with PBS for 5 min. Then, the slices were sealed using a fluorescence quencher containing DAPI for observation, and the expression of spinal dorsal keratins was detected by fluorescence microscopy and photographed for analysis.
Cell culture and treatments
BV-2 cells were resuscitated and adhered in DEME (HyClone, Utah, USA) medium containing 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin and streptomycin(Gibco), and incubated at 5% CO2, 37° until the logarithmic phase of cell growth was used for experiments. The purity of the required place in TNF-a (50 ng/ml, mice: CYT-252, Prospec, Rehovot, Israel) and Z-VAD (30 mM, G7232, Promega, Madison, WI) was co-induced for 24 h to establish an in vitro cellular necroptosis model. 25 Cells were pre-stimulated with GSK’872 (5 μM, Cat# 5.30389, Sigma, USA) and TLR4-IN-C34 (100 μM, Cat# SML0832, Sigma, USA) 2 h before the experiment. Two hours before experimentation.24,26 Cells were divided into four groups and subjected to the following treatments: Control group, untreated group; TNF-a group, TNF-a (50 ng/ml) only for 24 h; Z-VAD group, Z-VAD (30 μM) only for 24 h; and TNF-a+Z-VAD, TNF-a (50 ng/ml) and Z-VAD (30 μM) combined were induced for 24 h.
Western blotting
The L4–L6 segments of the spinal cord were removed, the cut tissue was homogenized in a RIPA lysis mixture containing 1% protease inhibitor. Protein concentration was determined using the BCA Protein Assay Kit; 20 µg of sample per well was added to SDS-PAGE gel and transferred to a PVDF membrane, which was closed with 5% skimmed milk powder at room temperature for 2 h, then incubated at 4℃ for overnight with primary antibodies against RIPK1 (1: 1000 dilution, ab300617, Abcam, USA), RIPK3 (1: 2000 dilution, ab255705, Abcam, USA), MLKL, IBA-1 (1: 500 dilution, ab178846, Abcam, USA), TLR4 (1:1000 dilution, AF7017, Affinity, USA), MyD88 (1:1000 dilution, AF5195, Affinity, USA), and GAPDH (1:10000 dilution, AF7021, Affinity, USA). After washing with Tris-buffered saline with Tween 20 (TBST) three times for 5 min, (HRP)-labeled goat anti-rabbit IgG antibody or goat anti-mice IgG antibody for 1.5 h at room temperature and washed three times with TBST. The bands were visualized using ECL enhanced chemiluminescence detection reagent. Relative band intensities were measured using Image J software and normalized by GAPDH.
Quantitative real-time polymerase chain reaction (qRT–PCR)
Total RNA was extracted from tissues and cells using TriQuick Reagent Total RNA Extraction Reagent, RNA concentration was determined using Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA) and RNA concentration was measured by Qubit (Life Technologies, Inc. Carlsbad, CA, USA) to measure RNA concentration. It was converted to complementary DNA (cDNA) using Hiscript III RT SuperMix Perfect for q PCR, selective programmed (50°C, 15 min/85°C, 5 s). Real-time PCR was performed using ChamQ SYBR qPCR Master Mix, Light Cycler 480 II. Fifteen microliter of PCR master mix was prepared as follows: 4.6 μl of c DNA template, 5 μl of 2 × Cham Q SYBR qPCR Master Mix, and 0.4 μl of gene-specific primers.
The PCR amplification procedure was as follows: 95°C, 5 min (stage 1: pre-denaturation), 95°C, 10 s, 60°C, 30 s (section 2: cycling reaction) for 40–50 cycles. During the qRT-PCR, the number of cycles for each sample for reach the set threshold signals were recorded. The relative fold change of the transcripts was calculated using the 2−△△ct method, and the endogenous GAPDH mRNA level were used for standardization (Table 1).
The sequences of primers used for qRT-PCR analysis of mRNA levels.

Statistical analysis
The GraphPad prism 9.4.1 system was used for statistical analysis of all experimental data. The data are expressed as the mean and standard error of the mean (SEM). Changes in the PWT were analyzed using two-way repeated measures analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test. When comparing the data between multiple groups, ANOVA tests are acceptable. If p < 0.05, the comparison is regarded as statistically significant.
Results
RIPK3-mediated necroptosis involved in neuropathic pain and GSK’872 attenuates neuropathic pain in CCI
The results showed that the mechanical and thermal pain thresholds on the CCI groups began to decrease on the third day after surgery, and this state of nociceptive hypersensitivity persisted until the 14th day after surgery. The necroptosis inhibitor GSK’872 was administered on the first postoperative day and for 14 days of intervention, and data from behavioral pain threshold measurements showed that inhibition of necroptosis alleviated pain sensitivity (Figure 2(a) and (b)).

GSK’872 alleviates neuropathic pain and inhibits RIPK3-mediated necroptosis. (a, b): GSK’872 attenuated both mechanical allodynia and thermal hypersensitivity Mechanical withdrawal threshold (MWT) was measured by the von Frey test, and thermal withdrawal threshold (TWL) was measured by thermal testing, each at different time points; n = 10 per group. (c–f): Western blot detection of RIPK1, RIPK3, and MLKL expression in spinal cord tissues of mice in each group; (g–i): qRT-PCR to detect the expression levels of RIPK1, RIPK3, and MLKL in the spinal cord tissues of mice in each group.
The potential effects of GSK’872, on CCI mice necroptosis were then investigated. Firstly, the protein levels of necroptosis-specific marker proteins RIPK1, RIPK3, and MLKL were analyzed in the spinal cord of groups of mice, including Sham, CCI, CCI + DMSO, and CCI + GSK’872 groups, using Western Blot. It was found that the levels of RIPK1, RIPK3, and MLKL were up-regulated in the CCI group compared with Sham. However, treatment with GSK’872 demonstrated an inhibitory effect on this elevated expression (Figure 2(c)–(i)). This suggesting that GSK’872 significantly inhibited RIPK3-mediated necroptosis in the spinal cord of neuropathic pain mice.
RIPK3 is involved in neuropathic pain by activating microglia polarization and GSK’872 inhibits microglia M1 polarization in mice neuropathic pain
To clarify which kind of cells is RIPK3 mainly expressed, mice were taken from the L4–L6 stage of the lumbar spinal cord at the end of the last behavioral tests, and the co-localization of RIPK3 was observed in neurons (NeuN), microglial cells (Iba-1) astrocytes (GFAP) at the dorsal horn of the spinal cord of the mice, respectively. The Immunofluorescence results showed that RIPK3 primarily expressed in Iba-1 cells (microglia) in the spinal cord (Figure 3(a) and (b)). We further examined the M1 and M2 microglia polarization markers in the spinal cord of mice. It could be observed that the expression of MI-type markers CD86 and iNOS was significantly higher in the CCI group compared to the Sham group. The expression of M2-type markers CD206 and ARG-1 was decreased considerably, and after the administration of GSK’872, the expression of MI-type markers CD86 and iNOS expression levels were down-regulated, and M2-type markers CD206 and ARG-1 expression levels were up-regulated after administration of GSK’872(Figure 3(c)–(f)), suggesting that RIPK3 be involved in neuropathic pain by activating microglia polarization.

RIPK3 expression was mainly in neuron and microglia and GSK’872 ameliorated microglia M1 polarization induced by neuropathic pain. (a): Double immunofluorescence labeling of RIPK3 with GFAP (an astrocyte marker), NeuN (a neuronal marker), or IBA-1 (a microglia marker) in the ipsilateral spinal cord dorsal horn of mice 14 days after CCI. (b): The quantification of cells with overlapping signals for the used markers, the expression of RIPK3 mainly in microglia and neuron. (c–f): GSK’872 reduced the expression of CD86 and iNOS mRNAs but increased the expression of CD206 and ARG-1 mRNAs.
RIPK3 regulate neuropathic pain by microglia polarization through the TLR4/MyD88 pathway
To further explored how RIPK3-mediated necroptosis regulates microglia polarization through the TLR4/MyD88 pathway. We used Western Blot and qRT-PCR to detect the changes in the TLR4/MyD88 pathway in the spinal cord of mice with CCI. After last behavioral tests, we took the lumbar segment of spinal cord tissues from the mice that the protein levels and mRNA expression levels of TLR4 and MyD88 were up-regulated in the CCI group compared with Sham group, and down-regulated after the application of necroptosis inhibitor GSK’872 treatment. These results suggest that the TLR4/MyD88 pathway is activated in CCI spinal cord mice (Figure 4(a)–(e)),suggesting that RIPK3 is involved in the regulation of neuropathic pain by microglia polarization through the TLR4/MyD88 pathway.

GSK’872 inhibits RIPK3/TLR4/MyD88 pathway involved in neuropathic pain. (a–c): Western blot detection of TLR4 and MyD88 expression in spinal cord tissues of mice in each group; (d, e): qRT-PCR detection of TLR4 and MyD88 expression levels in spinal cord tissues of mice in each group.
GSK’872 inhibits microglia M1-polarization and promote M2-type polarization through the TLR4/MyD88 pathway in BV2 cells
BV2 cells were stimulated by TNF-a (50 ng/ml) and Z-VAD (30 μM) for 24 h, then the cells were collected, and the expression of RIPK1, RIPK3, MLKL, and Iba-1 in microglia was detected by Western Blot and qRT-PCR. Compared with the Control group, the expression levels of RIPK1, RIPK3, MLKL, and Iba-1 were up-regulated in the TNF-a and Z-VAD combined induction group, indicating that the establishment of the necroptosis model was successful (Figure S1). Afterward, GSK’872 was given to observe the effect of necroptosis on the polarization regulation of microglia in vitro, and it could be observed that after the administration of GSK’872, the protein levels of RIPK1, RIPK3, MLKL, and Iba-1 were up-regulated in the model group compared to the Control group. It was also observed that the mRNA expression of microglia M1 markers CD86 and iNOS was down-regulated, the expression of M2 markers CD206 and ARG-1 was up-regulated. These results suggested that GSK’872 could modulate microglial polarization by regulating necroptosis (Figures 5(a)–(i) and 6(a)–(c)).

GSK’872 attenuated TNF-a+ Z-VAD- induced necroptosis in BV2 cells. (a–e): Western blot detected the expression of RIPK1, RIPK3, MLKL, and IBA-1 in BV2 cells in each group; (f–i): qRT-PCR detected the expression levels of RIPK1, RIPK3, MLKL, and IBA-1 in BV2 cells in each group.
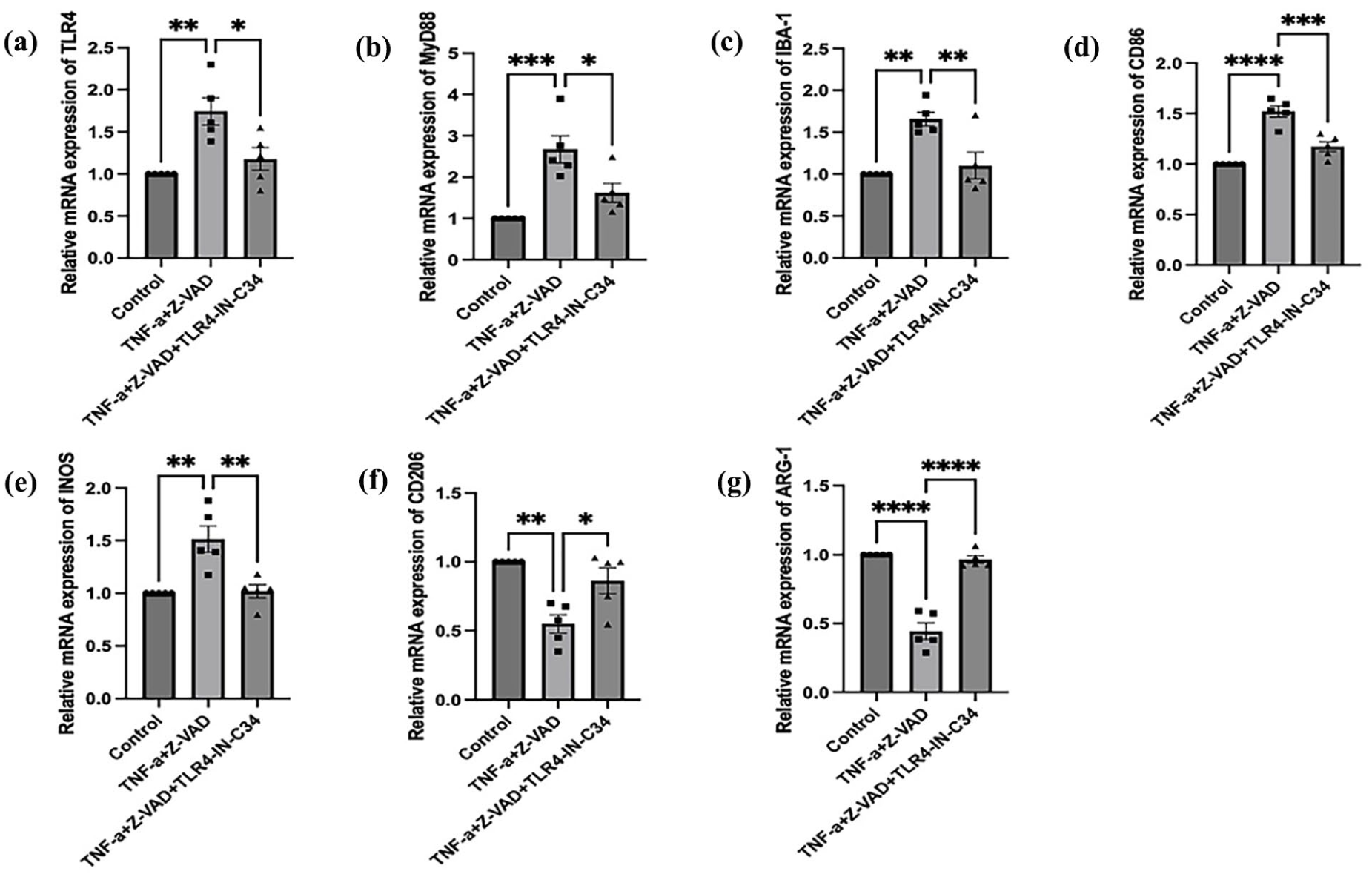
Inhibition of TLR4 attenuated TNF-a+ Z-VAD- induced M1 microglial polarization. (a–g): TLR4-IN-C34 reduced the expression of TLR4, MyD88, IBA-1, CD86, and iNOS mRNAs, but increased the expression of CD206 and ARG-1 mRNAs.
We further performed in vitro experiments to investigate the effect of necroptosis in BV2 cells polarization through the TLR4/MyD88 pathway. The results showed that TLR4 and MyD88 expression levels were up-regulated in the TNF-a and Z-VAD conduction group compared with the Control group (Figure 7(a)–(e)). It was observed that the mRNA expression of microglia M1-type markers CD86 and iNOS was down-regulated and the expression of M2-type markers CD206 and ARG-1 was up-regulated, suggesting that necroptosis may be able to modulate microglial polarization through the TLR4/MyD88 pathway (Figure S1). Afterward, we gave mice BV2 cells the TLR4 inhibitor TLR4-IN-C34 and pre-stimulated the cells with TLR4-IN-C34 (100 μM) 2 h before the experiment, after which BV2 cells were given a combination of TNF-a (50 ng/ml) and Z-VAD (30 μM) to induce the cells for 24 h. The TLR4, MyD88 pathways were detected in microglial cells by qRT-PCR, and mRNA expression of microglia M1-type markers CD86 and and M2-type markers CD206 and ARG-1. The results showed that the expression of TLR4, MyD88 and M1-type markers CD86 and iNOS was significantly up-regulated. The expression of M2-type markers CD206 and ARG-1 was significantly down-regulated, suggesting that the TLR4/MyD88 signaling pathway is involved in regulating necroptosis to modulate microglial polarization (Figure 6(d)–(g)).

GSK’872 attenuates TNF-a+ Z-VAD- induced necroptosis and M1 microglial polarization in BV2 cells via the TLR4/MyD88 pathway. (a–c): Western blot detects the expression of RIPK1, TLR4 and MyD88 in BV2 cells in each group; (d, e): qRT-PCR to detect the expression levels of TLR4 and MyD88 in BV2 cells in each group. (f–i): qRT-PCR to detect the mRNA expression of microglia M1-type markers CD86 and iNOS and M2-type markers CD206 and ARG-1 in each group.
we used TNF-a+ Z-VAD to establish an in vitro necroptosis model, the GSK’872 was given to observe the effect of necroptosis on the modulation of microglial cell polarization in vivo. It could be observed that, in comparison with the TNF-a+ Z-VAD group, the administration of GSK’872 group TLR4 and MyD88 protein levels, as well as mRNA expression levels was down-regulated. It was also observed that the mRNA expression of microglia M1-type markers CD86 and iNOS was down-regulated, and the expression of M2-type markers CD206 and ARG-1 was up-regulated. The above results indicated that GSK’872 could modulate microglial polarization through the TLR4/MyD88 pathway in vivo (Figure 7(a)–(i)).
Discussions
In our study, nerve injury caused changes in the sensory system, leading to neuropathic pain. Microglia are crucial in neuropathic pain development and maintenance. Our study found that the peripheral nerve injury activated the RIPK3/TLR4/MyD88 pathway and promoted M1-type microglial polarization. Inhibition of the TLR4/MyD88 pathway by a RIPK3 inhibitor reduced M1-type microglial polarization and ameliorated CCI-induced mechanical allodynia.
Receptor-interacting protein kinases (RIPKs) are key kinases in the necroptosis pathway, which is involved in inflammation, innate immunity, and signal transduction. The kinase activities of RIPK1 and RIPK3 activate the necroptosis pathway through necrosome formation and downstream MLKL protein activation, leading to organ damage, inflammation, chronic pain, and neurodegeneration. Our study found that in neuropathic pain, RIPK3 regulates necroptosis and microglial polarization and plays a key role in chronic pain-induced necroptosis. GSK’872 (RIPK3 inhibitor) prevents neuropathic pain by reducing pro-inflammatory cytokines and immune cells. 27 Zhang et al. 28 demonstrated that RIPK3 inhibitors prevent neuropathic pain by downregulating the RIPK1/RIPK3/MLKL signaling pathway. Our results demonstrated that RIPK3 expression is upregulated in CCI mice with neuropathic pain, suggesting RIPK3 inhibitors may as a novel therapeutic strategy for chronic pain by modulating necroptosis. Necroptosis and microglial polarization can influence disease progression, specifically, after the onset of the disease, inflammatory factors induce microglial cells to polarize to the M1 type, and the M1 cells can secrete TNF-a to kill oligodendrocytes, which then cause demyelinating injury.29,30 After spinal cord injury, the resulting chronic inflammation stimulates M1 polarization of microglia, and persistence of the M1 phenotype also promotes necroptosis of other cells via the TLR4/My88 pathway, necroptosis inhibitors are prevent neuropathic pain by inhibition of the TLR4/MyD88 pathway, which alters the polarization state of microglia.24,31. Our results showed that microglia were activated in the spinal cord of CCI mice after peripheral injury and participated in neuropathic pain.
The TLR4/MyD88 signaling pathway is closely linked to microglial polarization and is prominent in neuropathic pain and neurodegenerative diseases. 32 TLR4 activates NF-kB transcription via MyD88, releasing pro-inflammatory factors that drive microglia to M1-type polarization, exacerbating neurological damage. In certain pathological conditions, such as Alzheimer’s and Parkinson’s disease, activation of M1-polarized microglia exacerbates neuronal death and disease progression. Blocking TLR4 or downregulating MyD88 signaling attenuates the M1-type response, converting microglia to the M2-type and enhancing their repair function. For example, certain natural compounds (e.g., curcumin) and synthetic small molecule inhibitors have demonstrated the potential of inhibiting TLR4/MyD88 signaling to reduce both neuroinflammation and neuropathic pain. 33 Microglial polarization can cause neuroinflammation and retinopathy through activation of the TLR4 pathway. 17 Baicalin (a flavonoid) attenuates cognitive impairment and protects neurons from microglia-mediated neuroinflammation in mice by inhibiting the TLR4 pathway. 34 Cottonseed oil attenuates ischemic stroke injury by inhibiting microglial inflammatory activation via the TLR4 pathway. 35 Inhibitors targeting this pathway may be effective for treating neuropathic pain and related disorders.
RIPK3 is involved in necroptosis and interacts with TLR4 in various immune-related diseases. This finding is consistent with previous research. In liver disease, continuous activation of the TLR4 pathway leads to RIPK3 exacerbating inflammation and fibrosis through necroptosis.36,37 In renal diseases, RIPK3 attenuates ischemic reperfusion injury in rat kidney by interrupting the effect of indole sulfate on TLR4.38,39 RIPK3 affects transfusion-associated acute lung injury disease as well as acute pancreatitis through TLR4/MyD88.40,41 Our study demonstrated that inhibiting RIPK3/TLR4/MyD88 activity suppresses microglia M1 polarization. Using Western blot and qRT-PCR, we examined the expression of RIPK3, TLR4, MyD88, and microglia M1-type markers in the spinal cord of CCI mice. The results showed that the expression of RIPK3, TLR4, and MyD88 were significantly elevated, and administration of RIPK3 inhibitors were reduced. Future studies could further explore the specific mechanisms of this pathway and its interaction with other signaling pathways in neuropathic pain. The results suggesting that the RIPK3/TLR4/MyD88 pathway modulates microglia polarization to influence neuropathic pain progression in both vivo and in vitro.
In summary, our study demonstrates that the RIPK3/TLR4/MyD88 pathway plays a crucial role in microglial M1 polarization and neuropathic pain progression following peripheral nerve injury. However, intervening in necroptosis can regulate the state of microglial polarization, thereby alleviating neuropathic pain. These results highlight that RIPK3 regulates the M1/M2-polarization state of microglia via the TLR4/MyD88 pathway. This provides a new theoretical basis for understanding the pathogenesis of neuropathic pain and suggests a potential therapeutic target.
Supplemental Material
sj-docx-1-mpx-10.1177_17448069251377861 – Supplemental material for RIPK3 regulates microglial polarization through the TLR4/MyD88 pathway in neuropathic pain
Supplemental material, sj-docx-1-mpx-10.1177_17448069251377861 for RIPK3 regulates microglial polarization through the TLR4/MyD88 pathway in neuropathic pain by Sihan E, Qingbiao Song, Zhaokun Zhang and Yingxia Liang in Molecular Pain
Footnotes
Acknowledgements
We would like to thank the Laboratory of Anesthesia and Critical Care Medicine in Colleges and Universities of Shandong Province, School of Anesthesiology, Shandong Second Medical University, for providing research support.
Author contributions
YXL conceived and designed the study. SHE performed the experiments, ZKZ analyzed and interpreted the data. SHE and QBS wrote the first draft and revised the manuscript. YXL Edited the manuscript and Supervision, all authors read and approved the final manuscript.
Data availability statement
All data about this study are included in the article, and further inquiries can be directed to the corresponding authors.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China [Grant No. 82571407]and Shandong Provincial Natural Science Foundation [Grant No. ZR2025MS1396].
Supplemental material
Supplemental Material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.