
Abstract
Purpose:
This study aimed to determine whether serum-and glucocorticoid-inducible kinase1 (SGK1) activation-dependent histone deacetylase 4 (HDAC4) phosphorylation, nucleocytoplasmic trafficking, and subsequent regulation of high-mobility group protein box 1 (HMGB1) expression are involved in type 2 diabetic neuropathic pain (DNP).
Methods:
The type 2 diabetic neuropathic pain model was established in rats by feeding them with a high-fat and high-sugar diet for 8 weeks and then fasting them for 12 h, followed by a single intraperitoneal injection of streptozotocin (STZ, 35 mg/kg). SGK1 was inhibited in the spinal cord by intrathecal administration of the SGK1 inhibitor GSK-650394.
Results:
The present study revealed that pSGK1/tSGK1 was persistently upregulated in the spinal cord of rats with type-2 DNP. The downregulation of pSGK1/tSGK1 through the intrathecal injection of the SGK1 inhibitor GSK-650394 significantly ameliorated the pain hypersensitivity, relieved the abnormal expression of pHDAC4/tHDAC4 and HMGB1, and affected HDAC4 nucleocytoplasmic trafficking in DNP rats.
Conclusion:
Our data suggest that SGK1 in the spinal cord modulates type-2 DNP by regulating the HDAC4/HMGB1 pathway.
Introduction
Diabetes mellitus (DM) is one of the most common and rapidly increasing diseases worldwide. Approximately 90% of these patients have type 2 diabetes. 1 Diabetic neuropathic pain (DNP) is a common complication among patients with type 2 DM, and its characteristics include spontaneous pain, hyperalgesia, and allodynia. 2 The exact mechanisms of DNP are extremely complex, so they are difficult to treat.
Emerging research has revealed a strong connection between epigenetic modifications that govern pain-related gene expression and the emergence of neuropathic pain. 3 In epigenetic modulation mechanisms, histone acetylation is regulated by histone acetyltransferases and histone deacetylases (HDACs). 4 HDAC4, a member of class IIa HDACs, governs a transcriptional program critical for synaptic plasticity and memory. 5 Activation of HDAC4 and glucocorticoid receptor (GR) signaling has been demonstrated to play a vital role in stress-induced hyperalgesia in the medial prefrontal cortex of rats. 6 In addition, unlike other HDACS, an amazing feature of HDAC4 is that it can shuttle between the cytoplasm and nucleus, thereby affecting the expression of synaptic plasticity-related genes. 7
Serum-and glucocorticoid-inducible kinase1 (SGK1), a serine/threonine kinase, plays an important role in spinal nerve ligation (SNL)-induced nociceptive hypersensitivity through phosphorylation of HDAC4 to change its subcellular location, and pharmacological inhibition of SGK1 activity relieves HDAC4 cytoplasmic retention. 8 HDAC4 subcellular translocation depends on its signal transduction-related phosphorylation status; that is, phosphorylation causes HDAC4 to be retained in the cytoplasm, whereas dephosphorylated HDAC4 is imported into the nucleus, which is one of the mechanisms that regulate gene transcription. 9 Moreover, SGK1 has recently been reported to be associated with inflammatory and neuropathic pain, 10 revealing that SGK1 plays a crucial role in nociceptive hypersensitivity. However, it remains unknown whether SGK1 affects HDAC4 phosphorylation and nucleocytoplasmic trafficking, and whether this change regulates downstream gene transcription in a type 2 diabetic neuropathic pain model.
High-mobility group protein box 1 (HMGB1) plays an important role in the pathology of neuropathic pain caused by SNL and nerve injury. 11 SNL causes HDAC4 phosphorylation and cytoplasmic accumulation, which epigenetically alleviates HDAC4-suppressed hmgb1 gene transcription and increases HMGB1 expression. 12 In the present study, we hypothesized that the spinal cord SGK1-HDAC4-HMGB1 signaling pathway might be involved in type 2 DNP. The expression of total SGK1, pSGK1, total HDAC4, pHDAC4, and HMGB1, as well as HDAC4 nucleocytoplasmic trafficking, was first examined in the spinal cord of rats with type 2 DNP. The effect of suppression of spinal SGK1 through intrathecal injection of the SGK1 inhibitor GSK-650394 on HDAC4 phosphorylation, HDAC4 nucleocytoplasmic trafficking, HMGB1 expression in the spinal cord, and pain behaviors in type 2 DNP were also investigated.
Materials and methods
Animal preparations
Six-week-old healthy male Sprague-Dawley rats (n = 108), weighing 130–150 g, were provided by the Animal Experimental Center of Wenzhou Medical University. The rats were housed under a 12-h light/12-h dark cycle in an animal laboratory, with unlimited access to food and water, and the temperature was maintained at 23–25°C. All experimental and animal care procedures were reviewed and approved by the Animal Use Committee of Wenzhou Medical University, and the IASP’s ethical guidelines for pain research in animals were followed.
Type 2 diabetic neuropathic pain model and experimental grouping
The type 2 DNP models were established as described previously. 13 After 3 days of feeding adaptation, the rats were divided randomly into two groups: the control group (C group, n = 18) and the type 2 DM group (D group, n = 90). The D group was fed a high-sugar and high-fat diet compounded with the formula described previously for 8 weeks, and the C group was fed a normal diet as a control. After 8 weeks, the insulin sensitivity trial was performed by testing for fasting blood glucose and insulin levels in blood samples collected from the tail vein. Insulin sensitivity index (ISI) = 1/(fasting glucose × fasting insulin). 14 These results revealed a significant difference in ISI between the two groups (p < 0.05), implying that insulin resistance was successfully induced in the type 2 DM group. Subsequently, the D group rats were injected with freshly prepared STZ (35 mg/kg, Sigma Aldrich, St Louis, MO, USA) solution in 0.1 M citrate buffer intraperitoneally. In contrast, rats in the C group were intraperitoneally injected with the same dose of citrate buffer. Three days after STZ injection, rats in the D group with fasting plasma glucose ≥16.7 mmol/l in the tail vein were classified as type 2 DM rats. Comparing the thermal withdrawal latency (TWL) and mechanical withdrawal threshold (MWT) of these rats before and after 2 weeks of injecting STZ, the pain threshold (MWT and TWL) was reduced to 85% of the baseline threshold (before STZ injection), which is considered as a successful model of type 2 diabetic neuropathic pain (DNP). 15
After the DNP model was established successfully, the rats were assigned in a random, blinded manner to three groups (n = 18 per group): DNP group, DNP with the injection of GSK-650394 group (DNP+GSK group), DNP with the injection of vehicle (SC group). For these type -2 DM rats, neither TWL nor MWT >85% of the baseline threshold was defined as the painless group (PL group, n = 18). Since approximately 80% of type 2 diabetic rats developed type 2 DNP, 16 more rats were prepared to meet the requirements for the minimum number of animals per group.
Measuring insulin and calculation of the insulin sensitivity index
After fasting for 12 h, 1 ml of tail vein blood was collected and placed at 23–25°C in an animal laboratory for 30 min. After centrifugation (3000×g for 20 min), the insulin content of the supernatant was measured using an ELISA kit (Haixi Tang Biotechnology Co., Ltd., Shanghai, China). The insulin sensitivity index was calculated by the following formula: insulin sensitivity index = 1/(fasting glucose × fasting insulin). The natural logarithm was used to calculate these data because the insulin sensitivity index was not normally distributed.
Behavioral tests
To validate whether the type 2 DNP rat models were prepared successfully, behavioral tests were measured before STZ injection, on day 14 after STZ injection (as a reference for successful modeling) and on days 3, 7, and 14 after intrathecal injection, and the results were evaluated by a person blinded to the different groups.
For the Mechanical Withdrawal Threshold (MWT) test, rats were habituated in an organic glass box (22 × 22 × 22 cm) with a metal mesh (1 cm2) at the bottom for at least 15 min. After the accommodation period, the surface of the rear toes was stimulated vertically with the IITC 2390 series electronic von Frey tactile pain measurement instrument (Woodland Hills, CA, USA) to measure the MWT, with a single-stimulus duration of ≤1 s and a single-stimulus hold for ≤1 s. During the behavioral test, the intensity of the stimulus was recorded when rats lifted or licked their feet. The test was repeated five times and the MWT was the average of five measurements. 17
An IITC 336 plantar/tail flick analgesia meter (Woodland Hills, CA) was used to measure Thermal Withdrawal Latency (TWL). Rats were placed in a square, transparent, bottomless plexiglass box (20 × 15 × 30 cm) on a 3 mm thick glass plate. After a 15-min period of acclimatization period, the hind paw was exposed to thermal radiation. The rat lifted its foot and moved from the heat source, and the time from the onset of thermal radiation to paw withdrawal was recorded as TWL. A maximal cut-off time of 25 s was used to prevent unnecessary tissue damage. The hind paws of each rat were tested by a blinded observer five times at time intervals of 5 min. 18
Intrathecal catheter implantation
Animals were anesthetized with sodium pentobarbital (50 mg/ml), and a PE-10 catheter (Ningbo Science and Technology Park, The Software Technology Co., Ltd, Zhejiang, China) was inserted between the L4 and L5 vertebrae, with an additional 2 cm advance into the lumbar enlargement of the spinal cord. After implantation, the animals were allowed to recover for 5–6 days. To confirm catheter placement, 10 μl 2% lidocaine was administered through the catheter. A limb paralysis response without neurological deficits indicated that the catheter was positioned correctly. 19
Intervention of drugs
After successful implantation of the intrathecal catheter, the vehicle (1% dimethyl sulfoxide) and 200 nM/10μl of GSK-650394 (Tocris Bioscience, USA) were injected for 14 consecutive days, respectively. 8 Behavioral tests as described above were carried out on days 3, 7, and 14 after the injection.
Isolation of cytoplasmic and nuclear protein
Rats were immediately anesthetized after the behavioral tests. The spinal lumbar enlargement segment (L4-5 spinal cord, n = 4) was promptly removed, rapidly frozen, and stored in liquid nitrogen. Cytoplasmic and nuclear proteins were extracted from dorsal horn tissue by a Nuclear Extract Kit (Applygen Technologies Inc., Beijing, China). Tissue samples were accurately weighed, cut into small pieces with autoclaved scissors, and washed twice with phosphate-buffered saline (PBS). Usually, 50–100 μl of cytosol extraction buffer A (CEB-A) was required for every 10 mg of animal tissue according to the weight of the previously weighed tissue. The tissue was then ground on ice and transferred to pre-cooled 1.5 ml centrifuge tubes, shaken vigorously for 30 s, and incubated on ice for 10–15 min, during which time they were shaken for 15 s every 5 min. Depending on the amount of CEB-A added, 1/20 of the volume of cytosol extraction buffer B (CEB-B) was added, shaken for 10 s, incubated on ice for 1 min, and centrifuged at 1000×g for 5 min at 4°C. The pellet contained the crude nuclei. The supernatant was transferred to a new tube and centrifuged at 12,000×g for 5 min at 4°C. The supernatant is cytoplasmic protein. Next, pre-cooled nuclear extraction buffer (NEB) was added to the centrifuged precipitate at a ratio of 50–100 μl of NEB per 10 mg of tissue. The mixture was shaken for 15 s every 10 min and incubated on ice for 30 min. Finally, the samples were centrifuged at 12,000×g for 5 min at 4°C and the supernatants were collected as nuclear proteins.
Western blotting
Rats were immediately anesthetized after the behavioral tests. The spinal lumbar enlargement segment (L4-5 spinal cord, n = 6) was promptly removed and rapidly frozen in liquid nitrogen until further processing. The tissues were homogenized in lysis buffer (1:10wt/vol) comprised of 0.32 M of sucrose, 2 mM of ethylenediaminetetraacetic acid (EDTA), 10 mM of sodium phosphate, 2 mM of dithiothreitol, 1 mM of Phenylmethylsulfonyl fluoride, Protease and Phosphatase inhibitors (Sigma Aldrich), and 20 mM of Tris-HCL. After grinding and ultrasonication, the lysates were centrifuged at 12,000 rpm at 4°C for 25 min and the supernatant was collected. The protein concentration of the supernatant was measured using the bicinchoninic acid assay (BCA). Protein samples (30 μg/sample) were heated at 95°C for 10 min and separated by 10% SDS-polyacrylamide gel electrophoresis. These proteins were transferred onto polyvinylidene fluoride (PVDF) membranes using an electrophoretic transfer film instrument (Bio-Rad, Hercules, CA). The membranes were blocked with 10% nonfat milk in Tris-buffered saline-Tween (TBST) for 2 h at room temperature and subsequently incubated with primary antibodies at 4°C for 16 h. The primary antibodies used were as follows: rabbit anti-tSGK1(1:2000; Abcam, UK), rabbit anti-pSGK1 (Ser422,1:1000; Affinity, USA), rabbit anti-tHDAC4 (1:1000; Genetex, USA), rabbit anti-pHDAC4 (Ser632, 1:1000; Genetex, USA), mouse anti-HMGB1 (1:4000; Abcam, UK), anti-TBP (1:1500, EASYBIO, China), and anti-β-actin (1:3000, Bioworld, China). Then, the membranes were washed with TBST three times for 10 min each and incubated with goat anti-rabbit IgG (1:3000; Biosharp) or goat anti-mouse IgG (1:3000; Biosharp) for 2 h at room temperature. The protein bands were detected by enhanced chemiluminescence (ECL). Densitometric analysis of the Western blot bands was performed using Quantity One image analysis software.
Immunofluorescence
Rats (n = 4) were perfused with 200 ml of saline, followed by 300 ml of 4% paraformaldehyde (PFA) diluted in 0.1 M phosphate-buffered saline (PBS). Spinal lumbar enlargement segments were then isolated and fixed in 4% paraformaldehyde for 12 h, followed by dehydration in 30% sucrose for 3 days at 4°C. After 3 days, transverse spinal sections were cut at a thickness of 8 μm using a Leica cryostat. The spinal sections were washed thrice for 10 min in PBS, followed by treatment with 0.2% Triton X-100 in PBS for 30 min at room temperature. Non-specific binding was blocked with 10% donkey serum albumin in PBS (1 h). For double-labeling immunofluorescence, the sections were incubated overnight at 4°C with either (1) a mixture of rabbit anti-tHDAC4 (1:300; Abcam, USA) plus mouse anti-NeuN (1:200, Abcam, UK), mouse anti-GFAP (1:200, Santa Cruz, USA), or mouse anti-OX-42 (1:200, Santa Cruz USA), or (2) a mixture of rabbit anti-pHDAC4 (Ser632,1:200; Affinity, USA) and rabbit anti-pSGK1(Ser422,1:150; Affinity, USA). The secondary antibodies (goat anti-rabbit IgG or goat anti-mouse IgG at a dilution of 1:200; Invitrogen) were added to the slides, along with DAPI (Abcam, UK), and the mixture was incubated for 1 h at 37°C. Subsequently, the staining was examined using a fluorescence microscope (Leica, Germany).
Statistical analysis
All data in this experiment were analyzed using SPSS (version 20.0; SPSS Inc., Chicago, IL, USA) and are expressed as mean ± standard deviation (SD). Statistical comparison of the data was performed using one-way or two-way analysis of variance (ANOVA), or independent-samples t-test. When the ANOVA results exhibited a significant difference, pairwise comparisons between means were tested by the least significant difference method (LSD). Statistical significance was set at p-value < 0.05.
Results
Validation of type 2 diabetic neuropathic pain rat models
A rat model of type-2 DNP was established by providing a high-sugar, high-fat diet to elicit insulin resistance, in combination with a single intraperitoneal administration of STZ. After rats were fed a high-sugar, high-fat diet for 8 weeks, the body weight of the D group was significantly higher than that of the C group fed a normal diet (Figure 1(a), t = 9.144, p < 0.05). In addition, the rats in the D group had higher blood glucose levels (t = 9.449, p < 0.05). However, this did not meet the diagnostic criteria of T2DM (≥16.7 mmol/l), although rats in the D group had a higher insulin level (t = 25.135, p < 0.05) and lower insulin sensitivity index (Table 1, t = 11.188, p < 0.05). At 3 days after STZ injection, the blood glucose levels in diabetic rats were significantly higher than those in the C group, and this high blood glucose level was maintained until the end of the experiments (Figure 1(b), treatment, F(4,20) = 972.100, p < 0.05; time, F(4,20) = 525.500, p < 0.05; treatment × time, F(16,80) = 122.800, p < 0.05). The average blood glucose levels in the diabetic rats were higher than 16.7 mmol/l, indicating that type-2 diabetic models were successfully established.

(a–d) Comparison of body weight, blood glucose, thermal withdrawal latency (TWL), and mechanical withdrawal threshold (MWT) between groups (a) The comparison of body weight after a high-sugar, high fat diet or normal diet for 8 weeks. (C group: n = 18, D group: n = 90). *p < 0.05 versus C group. (b) The comparison of blood glucose before and 3 days after STZ injection and subarachnoid injection at 3, 7, and 14 days (n = 6). *p < 0.05 versus C group. (c) The comparison of TWL before and 14 days after STZ injection and subarachnoid injection at 3, 7, and 14 days (n = 6). *p < 0.05 versus C group, #p < 0.05 versus PL group. (d) The comparison of MWT before and 14 days after STZ injection and subarachnoid injection at 3, 7, and 14 days (n = 6). *p < 0.05 versus C group, #p < 0.05 versus PL group.
Comparison of blood glucose, insulin level, and insulin sensitivity index (ISI) after normal-diet (C group) or a high-sugar -fat diet (D group).

Data are shown as mean ± SEM (C group: n = 18, D group: n = 90).
p < 0.05 versus C group.
In addition, the DNP group showed significant decreases in TWL (treatment, F(4,20) = 53.270, p < 0.05; time, F(2,10) = 277.400, p < 0.05; treatment × time, F(8,40) = 49.640, p < 0.05) and MWT (treatment, F(4,20) = 12.89, p < 0.05; time, F(2,10) = 60.72, p < 0.05; treatment × time, F(8,40) = 11.78, p < 0.05) during the experimental period (STZ 14d-GSK-650394 14d) compared to rats in the C and PL groups (Figure 1(c) and (d)). These data indicated that neuropathic pain induced by type-2 diabetes was successfully established.
Increased phosphorylation of HDAC4 in DNP rats
To investigate the role of spinal HDAC4 in DNP, we first analyzed HDAC4 protein expression at different time points in DNP rats. We found that total HDAC4 (tHDAC4) was uniformly expressed in the DNP group on days 3, 7, and 14 after subarachnoid injection compared with the C and PL groups (Figure 2(a), F(4,25) = 1.523, p > 0.05). In contrast to tHDAC4 expression, a statistically significant increase in phosphorylated HDAC4 (pHDAC4) protein expression was observed in the DNP group compared to that in the C and PL groups at these time points (Figure 2(b), F(4,25) = 31.32, p < 0.05). These results indicated a potential functional association between pHDAC4 and DNP.

Expression of tHDAC4 and pHDAC4 proteins in the spinal cord of different groups. (a, b) Time-course analyses of tHDAC4 and pHDAC4 expression in the spinal cord of DNP rats (n = 6). pHDAC4 expression was significantly upregulated in the DNP rats on days 3, 7, and 14 after subarachnoid injection compared with the C and PL groups. *p < 0.05 versus C group, #p < 0.05 versus PL group, tHDAC4 were uniformly expressed at the corresponding time point, p > 0.05.
HDAC4 localizes in spinal neurons but not in astrocytes or microglia
Double immunofluorescence staining was performed to determine the type of cells that expressed tHDAC4 in the spinal cord obtained from the DNP group (on day 7 after subarachnoid injection). It was found that most of the tHDAC4 (green) was colocalized with NeuN (red) in the spinal cord dorsal horn instead of the microglial marker OX-42 (red) or the astrocytic marker GFAP (red) (Figure 3). These results indicated that DNP-associated spinal tHDAC4 was mainly present in dorsal horn neurons rather than in microglia or astrocytes.

Localization of tHDAC4 in the spinal cord (n = 4). tHDAC4 (green) was mainly colocalized with neuronal (NeuN, red) but not astrocyte (GFAP, red) or microglial (OX-42, red) markers. Thickness = 8 µm. Scale bar = 50 µm.
SGK1 inhibitor GSK-650394 attenuated SGK1, HDAC4 phosphorylation and pain hypersensitivity in DNP rats
As shown above, spinal cord pHDAC4 may contribute to the development of DNP. Given that SGK1 promoted HDAC4 phosphorylation in the SNL-induced neuropathic pain model, 8 alterations of SGK1 and HDAC4 expression in spinal cords obtained from the DNP and GSK-650394 groups (DNP+GSK group) were observed. Western blot indicated that the expression level of total SGK1 (tSGK1) remained unchanged (Figure 4(a–d), Day3, F(4,25) = 1.442, p = 0.249; Day7, F(4,25) = 0.992, p = 0.430; Day14, F(4,25) = 0.087, p = 0.986). Furthermore, the western blot analysis revealed that pSGK1/tSGK1 increased in the DNP group (Figure 4(a–e), *p < 0.05, #p < 0.05) when compared with rats in the C group or the PL group, while intrathecal injection of GSK-650394 effectively reversed the DNP-enhanced pSGK1/tSGK1 in the DNP+GSK group (Figure 4(a–c, e), Day3, F(4,25) = 15.775, p < 0.05; Day7, F(4,25) = 45.746, p < 0.05; Day14, F(4,25) = 13.936, p < 0.05) compared with the DNP group. Moreover, compared to the DNP group, intrathecal injection of GSK-650394 decreased the expression of pHDAC4 (Figure 4(a–c, f), Day3, F(4,25) = 14.667, p < 0.05; Day7, F(4,25) = 6.357, p < 0.05; Day14, F(4,25) = 12.988, p < 0.05). Daily administration of GSK-650394 alleviated DNP allodynia (TWL, treatment, F(4,20) = 110.100, p < 0.05; time, F(4,20) = 174.500, p < 0.05; treatment × time, F(16,80) = 39.060, p < 0.05. MWT, treatment, F(4,20) = 85.620, p < 0.05; time, F(4,20) = 56.430, p < 0.05; treatment × time, F(16,80) = 18.440, p < 0.05 ) on days 3, 7, and 14 after intrathecal administration (Figure 4(g, h)). These results indicate that intrathecal administration of GSK-650394 alleviates pain hypersensitivity, possibly through the inhibition of SGK1-dependent phosphorylation of HDAC4. This result suggests that SGK1-dependent phosphorylation of HDAC4 plays a vital role in the development of type-2 DNP.
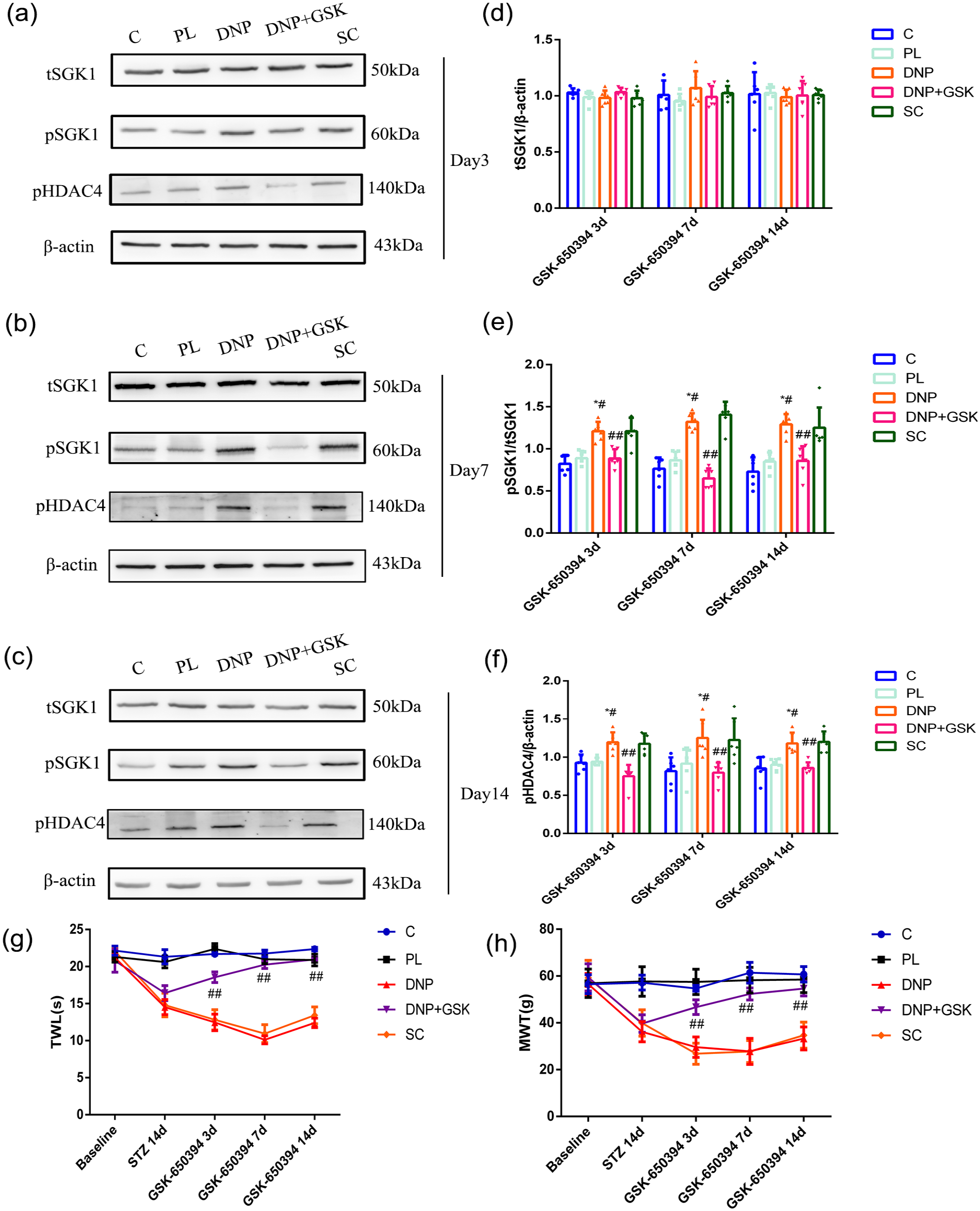
DNP induces phosphorylation of serum-and glucocorticoid-inducible kinase 1 (SGK1)-dependent histone deacetylase 4 (HDAC4). (a–f) Western blot analysis indicated that when compared with the C and PL groups, DNP enhanced the amount of pSGK1/tSGK1 and pHDAC4 on days 3, 7, and 14 after intrathecal administration, and administration of GSK-650394 decreased the DNP-enhanced pSGK1/tSGK1 and pHDAC4 expression. (n = 6). *p < 0.05 versus C group, #p < 0.05 versus PL group. ##p < 0.05 versus DNP group. (g, h) The SGK1 inhibitor alleviated the DNP-induced thermal withdrawal latency (TWL) and mechanical withdrawal threshold (MWT). (n = 6). ##p < 0.05 versus DNP group.
Co-localization of pSGK1with pHDAC4 in NeuN in type-2 DNP rats
Next, the double immunofluorescence staining revealed that pSGK1 (red) was colocalized with NeuN (green; Figure 5(a)). Given that tHDAC4 was colocalized with NeuN and pSGK1 (red) was colocalized with pHDAC4 (green; Figure 5(b)), it was concluded that pHDAC4 may directly bind to pSGK1.

pSGK1 colocalized with pHDAC4. (a) pSGK1 (red) was colocalized with NeuN (green), indicating that pSGK1 was expressed in NeuN (n = 4). (b) pSGK1 (red) was colocalized with pHDAC4 (green), indicating that HDAC4 may directly bind to SGK1. Thickness = 8 µm. Scale bar = 50 µm.
SGK1 inhibitor GSK-650394 prevented HDAC4 subcellular translocation and attenuated HMGB1 expression
Compared with the C and PL groups, we found that the cytoplasmic/nuclear ratio of tHDAC4 was significantly increased in the DNP group (Figure 6(a, b), *p < 0.05, #p < 0.05). While compared with the DNP group, intrathecal injection of GSK-650394 significantly reversed the cytoplasmic/nuclear ratio of tHDAC4 (Figure 6(a, b), F(4,25) = 35.325, p < 0.05). The shutting of HDAC4 is closely associated with HMGB1 expression. 12 DNP also increased HMGB1 expression in the spinal cord compared to that in the C and PL groups (Figure 6(c, d), *p < 0.05, #p < 0.05). Administration of GSK-650394 decreased the expression of HMGB1 (Figure 6(c, d), F(4,25) = 18.836, p < 0.05), consistent with the change in the cytoplasmic/nuclear ratio of tHDAC4.

HDAC4 subcellular translocation may increase HMGB1 expression to mediate DNP. (a, b) The HDAC4 subcellular translocation was enhanced in the DNP groups, whereas intrathecal injection of SGK1 inhibitor reversed this effect (n = 6). *p < 0.05 versus C group, #p < 0.05 versus PL group. ##p < 0.05 versus DNP group. (c, d) The expression of HMGB1 was elevated in the DNP group, whereas intrathecal injection of SGK1 inhibitor reversed this effect (n = 6). *p < 0.05 versus C group, #p < 0.05 versus PL group. ##p < 0.05 versus DNP group.
Discussion
The mechanisms underlying the development and maintenance of diabetic neuropathic pain are poorly understood. An increasing number of preclinical and clinical studies have revealed that neuropathic pain is closely associated with histone acetylation modifications, which regulate the expression of various pain-related genes.20–22 Histone acetylation is governed by two types of enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs). Histone acetylation is regulated by HATs, which acetylate lysine amino acids on histones to promote gene transcription. In contrast, HDACs that condense chromatin catalyze the removal of acetyl groups from histones, thereby reducing gene transcription. 23 Recently, emerging evidence has shown that HDAC inhibitors reduce hyperalgesia in animal models of neuropathic pain.24–27 These findings suggest that HDACs may be key regulators of the development of neuropathic pain. The results of this study provide evidence that HDAC4-associated epigenetic modification is an important mechanism through which DNP alters HMGB1 expression in the spinal cord.
SGK1 is a serine/threonine kinase that is highly expressed in the central nervous system (CNS), including the spinal cord. 28 Moreover, it is widely acknowledged that SGK1 plays a vital role in insulin resistance, and specific SGK1-knockout or SGK1 inhibitors improve glucose tolerance and insulin sensitivity, suggesting that SGK1 is a potential therapeutic target for metabolic syndrome and its associated consequences.29,30 Besides, SGK1 phosphorylation is enhanced during long-term potentiation (LTP), and transfection of inactive SGK1 impairs LTP in the hippocampus. 31 In addition, SGK1 phosphorylation has recently been linked to hyperalgesia, and pharmacological inhibition of spinal SGK1 activation relieves hyperalgesia.10,32 Therefore, we investigated whether SGK1 was involved in T2DM-induced neuropathic pain. In this study, we found that T2DM-induced pain hypersensitivity is accompanied by changes in SGK1 expression in the spinal cord. On days 3, 7, and 14 after subarachnoid injection, the expression of tSGK1 was unchanged, but the level of pSGK1/tSGK1 was significantly elevated in the DNP rats. Moreover, Pharmacological antagonism of spinal SGK1 activation by GSK-650394 ameliorated DNP-induced hyperalgesia and reduced the ratio of pSGK1/tSGK1. These results indicated that the phosphorylation of SGK1 in the spinal cord plays a vital role in the DNP.
Dysregulation of nucleocytoplasmic shuttling of HDAC4 is linked to various neurodevelopmental and neurodegenerative disorders.33,34 In addition, HDAC4 shuttles between the cytoplasm and nucleus, relying on the signal transduction-related phosphorylation status of HDAC4. 35 Moreover, HDAC4 activity can regulate synaptic protein expression, alter synapse shape, and modulate neuronal survival. 5 Increasing evidences revealed that phosphorylation-dependent cytosolic retention of HDAC4 in dorsal horn neurons contributes to the development of chronic pain.8,12,36 HDAC4 knockout decreases thermal hypersensitivity in the inflammatory pain induced by complete Freund’s adjuvant (CFA). 37 Knockdown of HDAC3, HDAC4, or both can increase the acetylation of foxo1, resulting in a decrease in the expression of gluconeogenic genes and an improvement in hyperglycemia in type-2 DM. 38 Several studies have shown that HDAC4 is closely related to the complications of diabetes, such as diabetic nephropathy, diabetic cardiomyopathy, and diabetic encephalopathy.39–41 However, whether HDAC4 plays a role in DNP remains unclear. In the type 2 DNP model, we found that a persistent increase in the levels of pHDAC4/tHDAC4 in the spinal cord of rats with DNP was associated with pain behavior. Increased expression of pHDAC4/tHDAC4 may be involved in sustaining DNP. However, the mechanism by which HDAC4 modulates pain remains unclear. SGK1 activation-dependent HDAC4 phosphorylation and 14-3-3β promotion of cytoplasmic HDAC4 retention in dorsal horn neurons play crucial roles in neuropathic pain maintenance. 8 This prompted us to investigate whether SGK1 activation-induced HDAC4 phosphorylation and trafficking were involved in T2DM-induced neuropathic pain. Our results suggest that DNP induced the phosphorylation of HDAC4 and facilitated cytoplasmic HDAC4 accumulation. Furthermore, intrathecal administration of the SGK1 specific inhibitor GSK-650394 decreased HDAC4 phosphorylation and cytoplasmic HDAC4 restriction. The main method of regulating the movement of HDAC4 between the nucleus and cytoplasm is through the phosphorylation of its serine residues. This leads to the export of HDAC4 from the nucleus, which recruits 14-3-3 to anchor the complex and restricts HDAC4 to the cytoplasm. 42 Furthermore, the localization of HDAC4 and SGK1 was explored, and the results demonstrated that tHDAC4 was mainly localized in neurons and that pHDAC4 colocalized with pSGK1 in the spinal cord of rats with diabetic neuropathic pain.
HMGB1 expression was increased in a spared nerve ligation (SNL) rat model, and injection of HMGB1 antibodies attenuated neuropathic pain. 43 In addition, recent research has shown that neuropathic injury promotes cytoplasmic HDAC4 accumulation, which uncouples HDAC4 from the hmgb1 gene to epigenetically alleviate HDAC4-suppressed HMGB1 transcription, resulting in increased HMGB1 expression in dorsal horn neurons. 12 The present study showed that the expression of HMGB1 is correlated with the expression of SGK1 and the phosphorylation/trafficking of HDAC4.
However, there are some limitations to our study. First, in addition to behavioral experiments, we also need additional experiments like electrophysiological experiment to comprehensively validate the effects of SGK1 inhibitors, which was not performed because of the technical constraints in our laboratory. Second, in this study, an intrathecal injection technique was used, but we cannot exclude that with the circulation of cerebral spinal fluid, pharmacological drugs may influence other neuronal tissues and induce some unknown effects.
In summary, we demonstrated that SGK1 modulates HMGB1 and pain behavior by influencing HDAC4 phosphorylation/trafficking in the spinal cord of DNP rats, which may provide a novel targeted drug for treating type-2 DNP.
Footnotes
Author contribution
Mao-Biao Zhang and Jia-Li Chen conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. Gai-Li Jai and Jia-Hui Lu designed the data collection instruments, collected data, carried out the initial analyses, and reviewed and revised the manuscript. Jun Li and Hong Cao coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Natural Science Foundation of China (Grant No. 81771487) and Scientific Research Foundation of Wenzhou (Grant No. Y20240723).