
Abstract
Recently, epigenetics involved in the regulation of gene expression has become a research hotspot. This study evaluated N4-acetylcytidine (ac4c) RNA acetylation in the spinal dorsal horn (SDH) of rats with cancer-induced bone pain (CIBP). The ac4C-specific RIP sequencing and NAT10-specific RIP sequencing were performed to identify the differences in ac4C acetylation and gene expression in the SDH between CIBP and sham groups, the relationship with the acetylation-modifying enzyme NAT10, and association analysis was performed. By interfering with the NAT10 expression, the relationship between some up-regulated genes and ac4C acetylation in CIBP was verified. In this study, we demonstrated that bone cancer increases the levels of NAT10 and the overall acetylation, inducing differential ac4C patterns in the SDH of rats. Through verification experiments, it was found that ac4C acetylation of some genes is regulated by NAT10, and differential ac4C patterns in RNA determine the expression of this RNA. We exposed that some CIBP-related gene expression was altered in the SDH of rats, which was regulated by differentially expressed ac4C acetylation.
Introduction
Cancer-induced bone pain (CIBP) is a common clinical pain due to bone metastasis. 1 Owing to the high incidence of CIBP, the quality of life of cancer patients is severely reduced, accompanied by a significant increase in the mortality rate.2,3 However, the current clinical treatment for CIBP is limited. Existing analgesic drugs have insurmountable toxic and side effects, which makes this problem particularly prominent. Additional studies are urgently required regarding the molecular mechanisms underlying CIBP to identify new therapeutic targets.
Epigenetics is essential in the occurrence and development of various disorders, affecting the structure and function of RNA through post-transcriptional modification is called epitranscriptomics, and has been one of the frontier research fields in biomedicine in recent years. 4 More than 150 types of natural RNA modifications have been reported, and previous studies have reported ac4C, m5C, m1A, and m7G-related content.5,6 N4-acetylcytidine (ac4C) RNA acetylation is a conserved chemical modification that acetylates N4-acetylcytosine under the action of RNA ac4C modifying enzymes. 7 In the field of protein covalent modification, many studies suggested that acetylation/deacetylation has become the second largest covalent modifier, closely related to pain and mainly affecting the level of post-transcriptional regulation.8,9 However, few reports have been on regulating chronic pain by RNA acetylation. RNA acetylation regulation has the potential to be a momentous breakthrough in the field of neuroscience research in the future, so it is of great significance to study it.
To prepare a CIBP model, Sprague Dawley (SD) rats were injected with 10 μL Walker-256 cancer cell suspension (1 × 106 cells) into the medial side of the left tibia. 10 An ac4C-specific RNA immunoprecipitation assay (acRIP-seq) of spinal dorsal horn (SDH) tissues of sham and CIBP rats was performed with high-throughput sequencing to investigate the differences in ac4C modification patterns between the sham and CIBP models and to additionally explore the function of ac4C alteration in the SDH of CIBP rats. Furthermore, 2552 differentially acetylated peaks within mRNA and 24 within lncRNAs were detected compared to SDH tissue. Intriguingly, genes encoding differentially acetylated peaks are involved in several essential biological pathways related to inflammation and pain transmission. Further investigation using bioinformatics analysis confirmed that the changes in ac4C in RNA were closely related to the expression of RNA by RIP-seq, quantitative real-time PCR (qPCR), and acRIP-qPCR methods. The current findings could provide novel insights into the mechanisms underlying CIBP.
Materials and methods
Animals
An SPF room was used to house the adult male Sprague Dawley rats (200 ± 20 g, n = 136) at 21–23°C with free access to food and water. The animal experiments were approved by the IACUC of Jiaxing University (JUMC2021-039) and conducted in according to the ethical standards of the Declaration of Helsinki and the guidelines for animal pain research of the International Association for the Study of Pain.
CIBP model establishment and organization collection
The CIBP model establishment and organization collection took place as previously described. In brief, SD rats were administered with Walker-256 breast cancer cell suspensions (10 μL, 1 × 106/mL) or heat-killed cells (sham group) into the medial side of the left tibia. For subsequent analyses, liquid nitrogen was used to store the rapidly collected L4–L6 left enlargement of the SDH from CIBP and sham rats. Detailed methods are listed in Supplementary Method S1.
Mechanical paw-withdrawal threshold (MWT) test
The MWT was tested as previously described. 11 The methods used are listed in Supplementary Method S1. MWT (g) was recorded.
CATWALK automated gait analysis
Gait analysis was carried out using the CATWALK XT system (AsterWee Information Technology), proving that the system is a reliable method for recording and analyzing the voluntary movements of rats in closed walkways, representing a trustworthy way to measure pain-related behaviors.12,13 Gait analysis was carried out as outlined previously.12,14 The maximum contact area, maximum contact intensity, and mean intensity were identified to evaluate the dynamic behavior related to CIBP. The detailed methods and definitions of the maximum contact area, maximum contact intensity, and mean intensity are listed in Supplementary Method S1. All data were calculated as percentages of the ipsilateral (left) and contralateral (right) hind paws.
Three-dimensional (3D) computed tomography (CT) reconstruction
A 3D CT bone reconstruction method was used to observe the destruction of the bone. The rats were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg) before undergoing CT tomography. According to a previous study, 15 the 3D CT reconstruction is described in Supplementary Methods S1.
Histochemical staining
Histochemical staining was performed as previously described. 15 Supplementary Method S1 illustrates the detailed methods.
RNA preparation
Three biological replicates were chosen for each group, followed by extracting total RNA from the tissue following the manufacturer’s instructions. We utilized an EpiTMac4Cimmunoprecipitation kit (Epibiotek, cat. no R1815), added RNA interruption buffer, incubated at 70°C for 6 min. It was followed by the immediate addition of EDTA to stop the reaction, we purified and recovered the fragmented RNA with Zymo RNA Clean and Concentrator-25 Kit (Zymo Research, Cat. No R1017).
Acetylated RNA immunoprecipitation sequencing and RNA sequencing
Anti-ac4C antibody (1:50, rabbit, Abcam) and washed protein G-magnetic beads were mixed in precipitation buffer, followed by 4–6 h incubation at 4°C. After separation of the magnetic beads, the supernatant was removed and pelleted. Then, we washed the buffer twice succeeded by removing the supernatant and adding segmented RNA, 5× precipitation buffer together with RNase inhibitor, and after overnight reaction at 4°C, RNAs were washed with both low- and high-salt precipitation buffers twice. The product was recovered using the HiPure Cell miRNA Kit. We used EpiTM mini longRNA-seq kit (Epbiotek, cat. no E1802) to prepare the library. Random primers, TSO, and reverse transcriptase were added to the recovered product, reacted at 40°C for 90 min, and at 70°C for 15 min to inactivate the enzyme, according to the smart principle synthesized first-strand cDNA. After magnetic bead purification, 2xpfuMax HiFi PCR ProMix and index primers for PCR amplification and enrichment of library fragments, magnetic bead purification and screening fragments, secondary PCR amplification and enrichment reaction on the obtained fragments, and DNA purification magnetic bead library fragments were performed. Using the BiopticQsep100 Analyzer, the quality inspection of the library was performed, and the high-throughput sequencing platform of NovaSeq was used for sequencing in the PE150 sequencing mode.
Sequencing data were analyzed, including peak calling analysis, peak visualization, peak statistical analysis, basic characteristics of ac4C, differential peak analysis, motif analysis, RNA-seq expression differences, NAT10-specific RIP-seq difference, ac4C modification difference, and association analysis of differential gene GO and KEGG analysis.
NAT10-specific RNA immunoprecipitation and sequencing
Using 1 mL IP lysis buffer, the total RNA of tissue was extracted, lysed on ice, or rotated at 4°C for 30 min and succeeded for 10 min at 12000×g centrifugation and the supernatant was transferred to a new 1.5 mL EP tube. Moreover, 10% of the lysate volume was used to extract RNA as an input sample and its integrity was checked. Further, 40 μL protein G magnetic beads were removed, washed with IP buffer and lysate, followed by the addition of antibody against NAT10 (1:50, rabbit, Abcam), and incubated at 4°C for a whole night. Then, the supernatant was transferred to a new 1.5 mL EP tube. The magnetic bead mixture was transferred onto a magnetic rack, the supernatant after clarification was discarded, 1 mL of wash buffer was added, mixed well at 4°C, rotated for 10 min, and the washing was repeated thrice. 1 mL of TRIzol™ Reagent (Invitrogen™, cat. no15596018) was added, and RNA extraction was performed using the phenol-chloroform method. The RNA obtained from the sample was isolated, and the library was prepared using the EpiTM mini longRNA-seq kit. After removing the ribosomal RNA, random primers, TSO, and reverse transcriptase were added, performing the reaction at 40 and 70°C for 90 and 15 min, respectively, to inactivate the enzyme, according to the smart principle to synthesize the first-strand cDNA After magnetic bead purification, there was an addition of 2xpfuMax HiFi PCR ProMix and index primers to PCR amplification and enriched the library fragments. The magnetic beads were used to screen the fragments and perform a secondary PCR amplification and enrichment reaction on the obtained fragments and DNA purification of the magnetic bead library fragments. Using the BiopticQsep100 Analyzer, libraries were quality checked by matching their size distribution to the theoretical size. The NovaSeq 6000 high-throughput sequencing system was used in the PE150 sequencing mode.
Gene-Specific acetylated RNA immunoprecipitation qPCR
The RNA was bound with anti-ac4C antibody (1:50, rabbit, Abcam) and proteinase A agarose affinity matrix overnight at 4°C. Moreover, 100 μg total RNA in co-immunoprecipitation buffer (50 mm Tris HCl, 750 mm NaCl, and 0.5% Igepal CA-630) containing RNase inhibitors were incubated at 4°C for 3 h, washed with proteinase K containing Debuff (5 mm Tris·HCl, 1 mm EDTA, and 0.05% SDS) to elute RNA from the beads. RNA ac4C was purified and extracted using phenol/chloroform. RNA primers (Table S1) were designed and prepared and qPCR was performed.
Screening siRNA sequence targeting rat NAT10
Supplementary Method S1 describes the scrambled and NAT10 siRNA001, siRNA002, and siRNA003 sequences and knockdown experiments.
qPCR
Supplementary method S1 describes the qPCR method and analysis.
Western blot
Supplementary method S1 describes the Western blot method and analysis.
Statistical analysis
For acRIP-seq data processing, we utilized Cutadapt (v2.5); adapters were trimmed, and sequences were filtered, along with the alignment of the remaining reads to the human Ensemble genomeGRCh38 (mouse Ensemble genome GRCm38) by the Hisat2 aligner (v2.1.0) under parameters: “--rna-strandness RF. “ Using the exomePeak R package (v2.13.2), the ac4C peaks were identified under parameters: “PEAK_CUTOFF_PVALUE = 0.05, PEAK_CUTOFF_FDR = NA, FRAGMENT_LENGTH = 200”. Employing the exomePeak R package, differential ac4C peaks were identified under parameters: “PEAK_CUTOFF_PVALUE = 0.05, PEAK_CUTOFF_FDR = NA, FRAGMENT_LENGTH = 200”. GO and KEGG analyses were conducted using cluster profile R package (v3.6.0). Visualization of ac4C-RNA-related genomic features was performed using the Guitar R package (v1.16.0). Detected ac4C peaks with a p-value <0.05, set for the de novo motif analysis by HOMER (v4.10.4) under the following parameters: ‘-len 12 -rna.’
Data were presented as mean ± SEM, and calculations were performed using SPSS version 20.0. One-way repeated measures analysis of variance followed by the Bonferroni test was conducted to analyze the differences in the molecular expression or behavioral scores among groups, respectively. p-values <0.05.
Results
CIBP model verification
The CIBP rat model was established by inoculating Walker 256 breast cancer cells into the tibial bone marrow cavity, and four methods were used for verification: CatWalk gait analysis, MWT test, CT 3D imaging, and hematoxylin staining.
Evaluating gait parameters using CatWalk gait analysis is an objective approach for assessing spontaneous pain behaviors due to bone cancer.
16
The chosen parameters with significant alterations in SD rats were as follows: (1) maximum contact area, (2) maximum contact maximum intensity, and (3) mean intensity to display pain-related behaviors. CatWalk gait analysis was conducted on day 9 after injection of Walker 256 breast cancer cells. As shown in Figure 1(a) and (b), in sham rats, there was an approximately 100% left/right hind paw ratio in the three chosen parameters, which showed a significant decrease following tumor inoculation, representing CIBP behaviors from different aspects (∗p < 0.05, ∗∗p < 0.01 vs. the sham group; n = 6, one-way ANOVA). Mechanical hyperalgesia was assessed on days 3, 6, 9, and 12 using the MWT. Baseline measurements were taken 1 day prior to the operation. We measured the MWT of the ipsilateral hind paws. The data revealed a downward trend in the CIBP group on days 6–12 than the sham (∗p < 0.05 and ∗∗p < 0.01 vs. the sham group; ##p < 0.01 vs. the baseline, n = 8, repeated-measures (ANOVA, Figure 1(c)). Harvesting the operating hind paw for a CT scan, as well as for hematoxylin-eosin staining, was performed when the behavioral assessment ended. The images indicated severe erosion of sclerotin and cancer cells that filled the marrow cavity (Figure 1(d) and (e)), suggesting the successful establishment of the CIBP rat model. The successfully established CIBP model. (a) Representation of the CatWalk gait findings, including print and timing view, and the print intensity in both groups. LH: left hind paw; RH: right hind paw. (b) The percentages of the ipsilateral (left)/contralateral (right) hind paw of the three chosen parameters showed a significant decrease in CIBP rats compared to the sham rats. (∗p < 0.05 and ∗∗p < 0.01 vs. the sham group; n = 6, one-way ANOVA). (c) The threshold of the mechanical pain of the hindfoot in CIBP rats reduced from day 6–12 following the surgery (∗p < 0.05 and ∗∗p < 0.01 vs. the sham group; ##p < 0.01 vs. the baseline, n = 8, repeated-measures ANOVA). MWT: mechanical paw-withdrawal threshold. (d) 3D CT reconstruction revealed the significant destruction of the cortical bone on day 12 following tumor inoculation. Arrowhead indicates the spot with abnormal bone structure due to tumor cells. (e) Hematoxylin-eosin staining demonstrates the lymphocytes, red blood cells, and macrophages that fill the bone marrow cavity of sham rats, and the observed regular arrangement of trabecular bone within it. The appearance of cancer cells (dotted lines) and bone resorption pits (yellow arrows) in the bone marrow cavity of CIBP rats following tumor inoculation. Scale bars: 50 μm (bottom row).
Bone cancer alters SDH ac4C-modifying enzyme NAT10 expression
The lumbar spinal cord enlargements (L4-L6) were collected to assess the NAT10 expression in the SDH tissue at 3, 9, and 15 days after the successful establishment of the CIBP model. We performed qPCR among the total RNA of SDH tissue from CIBP and sham rats. First, we found a significant increase in mRNA expression of NAT10 (Figure 2(a)) at postoperative days 9 and 15 in CIBP rats. Meanwhile, protein expression levels of NAT10 were also increased in SDH tissue from CIBP rats at postoperative days 9 and 15 (Figure 2(b) and (c)). NAT10 expression is increased in the SDH tissue at 9, and 15 days after the successful establishment of the CIBP model. (a) mRNA expression of NAT10 in SDH from CIBP rats was increased compared with expression in their corresponding sham, as determined by q PCR. (b) The protein level of NAT10 in SDH from CIBP rats was increased compared with expression in their corresponding sham, as determined by Western blotting. (c) Densitometry quantitation of (b). All data are expressed as the means ± SE. *p < 0.05, **p < 0.01 (n = 3 independent experiments).
General characteristics of ac4C acetylation in SDH tissue of sham and CIBP groups
The acRIP-seq analysis of RNA from SDH tissue of SD rats revealed 1514 and 14 non-overlapping ac4C peaks in 1342 coding gene transcripts (mRNAs) and 13 long noncoding transcripts (lncRNAs), respectively, in the sham group, and 1814 and 11 non-overlapping ac4C peaks in 1598 mRNAs and 11 lncRNAs, respectively, in the CIBP group. Of these, 365 peaks (only 12.32% in both groups) overlapped between the sham and CIBP groups (Figure 3(a)). The lower percentage of overlapping ac4C peaks in the mRNA indicated differences in the ac4C patterns between the two groups. The N4-acetylcytidine acetylation overview in mRNAs of sham and CIBP groups. (a) Venn diagram presenting the overlap of ac4C peaks within mRNAs in both groups. (b) The proportion of genes harbor various ac4C peaks in the sham and CIBP groups. Most genes harbor only one ac4C peak. (c) Pie charts displaying the percentage of ac4C peaks in six non-overlapping segments of transcripts. The ac4C peaks were most enriched in the CDS and 3′ UTR. (d) Distributions of fold enrichment of ac4C peaks in six segments. The mean fold enrichment in the exon segments was the largest in the sham group, while that value in stop codon segments was the largest in the CIBP group. Error bars indicate the mean standard error. ac4C: N4-acetylcytidine.
Notably, 89.47% of the ac4C acetylation-encoding genes in the sham group and 88.16% of the ac4C acetylation-encoding genes in the CIBP group had only one ac4C peak, whereas relatively few genes contained two or more (Figure 3(b)).
To evaluate the distribution profile of ac4C peaks in mRNAs, there were six transcript fragment divisions:5′ UTR, start codon fragment (400 nucleotides on the center of the start codon), coding sequence (CDS), exon, stop codon fragment (400 nucleotides at the center of the stop codon), and 3′ UTR. ac4C was found to be the most frequently contained in the CDS and 3′ UTRs (Figure 3(c)). Furthermore, in the sham group, the ac4C peak showed an increased fold enrichment in the stop codon segment (Figure 3(d), left panel). However, in the CIBP group, exons had higher fold-enriched peaks (Figure 3(d), right panel), indicating a differential ac4C pattern of bone cancer induction in the SD rat dorsal horn tissue.
Distribution of differentially acetylated ac4C sites
General numbers of differentially acetylated peaks and associated genes.

Top 10 upacetylated peaks.
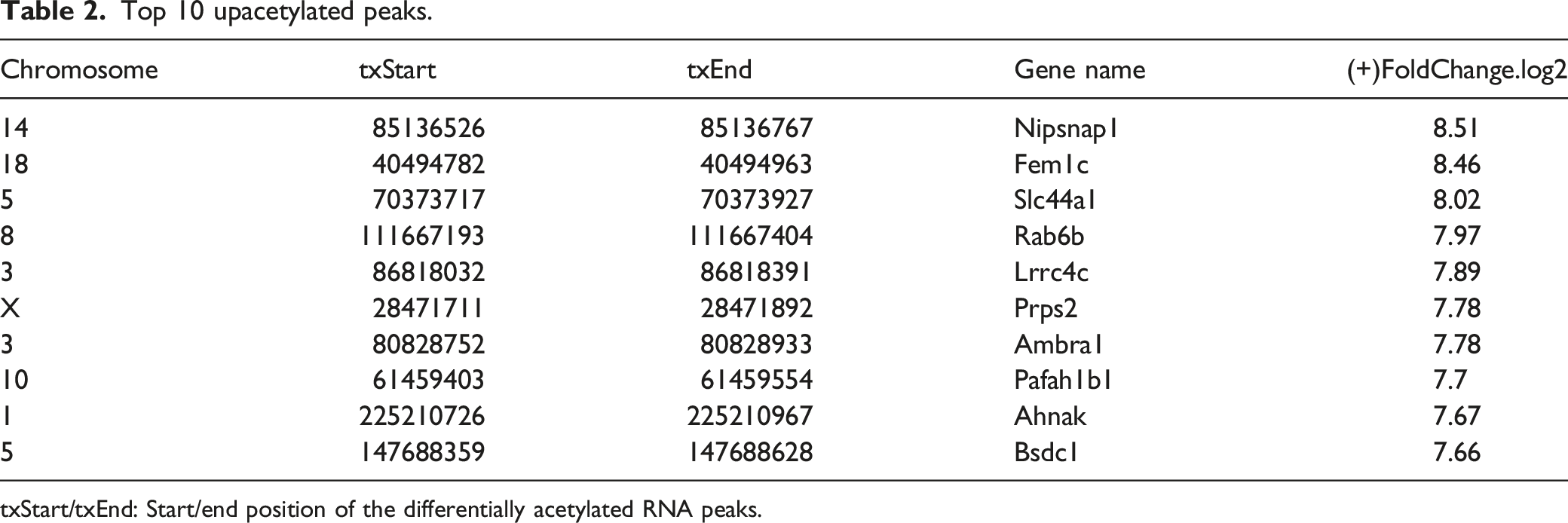
txStart/txEnd: Start/end position of the differentially acetylated RNA peaks.
Top 10 downacetylated peaks.

txStart/txEnd: Start/end position of the differentially acetylated RNA peaks.
Mapping of all DAASs in mRNAs and lncRNAs to chromosomes was performed to make their distribution profiles accessible (Figure 4(a)). The top three chromosomes that harbored the most DAASs were chromosomes 1 (112), 10 (73), and 3 (64). Nevertheless, when normalizing the number of DAASs harbored by chromosomes via the length of the respective chromosomes, the top three chromosomes with the highest relative DAAS densities were 1, 2, and 3 (Figure 4(b)). Additional analysis revealed that most of the DAAS in the mRNA were within the CDS and 3′-UTR (Figure 4(c)). In addition, the 3′UTR, 5′UTR, CDS, and sites within the exons all had high fold changes with little difference in both the upper and lower acetylation sites (Figure 4(d)), revealing genome-wide acetylation and deacetylation preferences in the CIBP group compared to the sham group. Distribution of differentially acetylated N4-acetylcytidine sites. (a) Chromosomal distribution of all DAA sites within RNAs. (b) Relative occupancy of differentially acetylated ac4C sites in each chromosome was normalized by the length of the respective chromosome. (c) Pie chart displaying the percentage of DAA peaks in four non-overlapping segments. (d) Statistics of the fold change of DAA peaks in four segments. The upper heatmap and the lower histogram represent the distribution and the mean of the fold change, respectively. Error bars indicate the mean standard error. DAA: Differentially acetylated N4-acetylcytidine.
Screening the associated genes
The acetylation-modified genes were correlated with the expression changes of the target genes, and a group of CIBP-related genes was screened for further research. In this study, we found 627 genes and their ac4C peak expression increased (Figure 5(a)), and 510 genes and their ac4C peak expression decreased (Figure 5(b)) in the CIBP model. The RNA ac4C-modifying enzyme (NAT10) is the enzyme used for ac4C. RIP-seq experiments were performed to analyze the interaction between RNAs and NAT10, 8671 genes were significantly enriched in NAT10 immunoprecipitated. The acRIP-seq analysis found a total of 821 genes with ac4C peak fold change >2 or <0.5 (CIBP vs sham), of which 91.35% (750/821) appeared in the RIP-seq results. Correlations were found between changes in gene expression in the SDH of CIBP rats and NAT10-mediated acetylation modification. In summary, both RNA and its acetylation expression were up-regulated, and RIP−target NAT10 (log2FC) ≥ 1 condition from the many genes detected in the SDH tissue of rats with CIBP, 209 were chosen for further analysis because they were closely related to the disease (Figure 5(a)). Association analysis of differentially expressed genes, differential ac4C peak, and RIP-target NAT10. (a) The expression of ac4C peak and gene expression were both up-regulated. The concentration of red dots represents the amount of RNA binding to NAT10. (b) The expression of ac4C peak and gene expression were both down-regulated.
Bioinformatics analysis of associated genes
To determine the function of ac4C in CIBP rats, GO and KEGG analyses were executed on the associated genes. GO analysis was performed on the related genes to study their functions, Moreover, biological process (BP) analysis findings showed that the associated genes were mostly involved in the hindbrain and limbic system development, and negative regulation of angiogenesis. Molecular function (MF) analysis indicated that most of these genes correlated with nucleoside-triphosphatase, regulatory activity, and GTPase regulatory activity. Cellular component (CC) analysis exposed that the gene distribution was mainly in the growth cone, polarized growth sites, and cell fronts (Figure 6(a)). Furthermore, the KEGG pathway revealed the relationship of most of these genes to the pathways of neurodegeneration, including multiple diseases, RNA degradation, tight junctions, protein processing in the endoplasmic reticulum, Parkinson’s disease, aldosterone-regulated sodium reabsorption, amyotrophic lateral sclerosis, cGMP-PKG signaling pathway salivary secretion, and cell adhesion molecules (Figure 6(b)). GO and KEGG analyses of coding genes that harbor differentially acetylated N4-acetylcytidine sites (cont.). (a) The top GO terms of BP, CC, and MF had a significant enrichment for the candidate key genes. (b) The bar plot demonstrates the top 10 enrichment scores of the significant enrichment pathway for the candidate key genes.
Validation of candidate genes
Using the current understanding of CIBP pathogenesis and a comprehensive bioinformatics analysis of associated genes, 15 candidate genes were screened for verification. In the verification experiment, siRNA sequences targeting the ac4C-modifying enzyme NAT10 were screened for NAT10 expression reduction in the SDH of the rats (Figure 7(a) and (b)). Additionally, qPCR and acRIP-qPCR were used to detect changes in the expression of candidate genes and their ac4C peaks in SDH tissues of CIBP rats, and the effect of inhibiting the ac4C modification enzyme NAT10 on the expression of these genes. The experiment was divided into six groups: sham, CIBP 3-days, CIBP 9-days, CIBP 15-days, CIBP 15-days MC-siRNA, and NAT10-siRNA (intrathecal injection of NAT10-siRNA in CIBP rats). As shown in Figure 7(c), among the 15 validated candidate genes, nine genes were overexpressed in the CIBP group and suppressed in the NAT10-siRNA group. Validation of candidate genes. (a) The expression of NAT10 showed a significant reduction by Western blot analysis in CIBP rats SDH transfected with NAT10-siRNA (10 nmol, i.t.) than the counterparts transfected with mismatch siRNA (MC-siRNA, 10 nmol, i.t.). GAPDH represents a loading control. The data are reported as the mean ± SEM of three rats per group. **p < 0.01 vs. MC-siRNA. (b) Densitometry quantitation of (a). (c) Heatmap of 15 candidate genes and their differential expressed ac4C peaks by qPCR and acRIP-qPCR among the sham group, CIBP 3d group, CIBP 9d group, CIBP 15d group, CIBP 15d + MC-siRNA group and CIBP 15d +NAT10-siRNA group (n = 3).
Seven ac4C-modified genes were overexpressed in the CIBP group and were suppressed in the NAT10-siRNA group. Correlation analysis was performed on genes with differential expression and ac4C peaks. It was found that seven genes were positively correlated, namely Ambra1, Cnot1, Gabbr1, Nfat5, Pafah1b1, Srgap3, and Nrxn2. The research revealed the altered expression of certain genes in the SDH of CIBP rats. This change was regulated by ac4C acetylation. These findings provide new clues to study the occurrence and development mechanism of CIBP, providing potential targets to treat CIBP.
Discussion
The 2020 Global Cancer Report reported an increase of 19.29 million new cancer cases and 9.96 million cancer-related deaths in 2020, respectively. 17 Most cancer patients experience pain, and cancer pain is higher in metastatic or end-stage patients, 38.0% of cancer patients report moderate to severe pain. 18 According to reports, the incidence of bone metastasis in patients with advanced breast and prostate cancers is as high as 90%. 19 It is evident that many patients are affected by CIBP. Although there are many treatment methods for CIBP, including drug therapy and minimally invasive interventional therapy, however, there are many patients with ineffectively controlled cancer pain, negatively impacting their quality of life.20,21 The pain of patients urges scholars to conduct continuous and in-depth research on CIBP to explore its pathogenesis and discover new analgesic drug targets and mechanisms of action.
With the extensively studied epigenome, creating an “epitranscriptome” via chemical alteration of ribonucleosides extends the regulatory content inherent in mRNAs. 22 More than 140 ribonucleoside modifications occur in all four nucleobases, and have been reported in prokaryotes, archaea, and eukaryotes. 23 Despite the regulatory potential due to the diversity of modified residues in RNA, extensive investigation has significant challenges due to the limited accessibility of reagents and poor mechanistic awareness. Most studies have focused on precisely examining abundant transfer RNAs and ribosomal RNAs; however, what is known about modifications within mRNAs remains inadequate. Upon examination, mRNA modifications were found to affect post-transcriptional metabolism by regulating mRNA’s stability, processing, and translation ability. 22
By focusing only on cytidine, 11 base alterations were identified in RNA; only three were conserved in all domains of life:5-methylcytidine (m5C), 5-hydroxymethylcytidine (hm5C), and ac4C. 23 Among them, direct analogs of m5C and hm5C are found in DNA, and current knowledge of DNA methylation has facilitated the study of their regulation, distribution, and function in RNA.24–26 In contrast, ac4C remains uninvestigated. Originally described in the bacterial tRNAmet anticodon, 27 ac4C has been identified in eukaryotic serine and leucine transfer RNAs and 18S ribosomal RNAs (rRNAs). 23 In all cases, the NAT10 enzyme and its homologs catalyze ac4C production.28–30 Recently, unbiased mass spectrometry (MS) studies have increased the probability that the NAT10/ac4C axis extends to polyadenylated (poly(A)) RNAs. The proteomic features of the mRNA-interactome indicated that NAT10 is a poly(A)-interacting factor, and ac4C was constantly identified in liquid chromatography (LC)-MS/MS of poly(A) RNA isolated from various human cell types at a level comparable to the 5′7-methylguanosine (m7G) cap.31,32 These results suggested that ac4C is present in the mRNA at physiologically relevant levels.
We used a transcriptome-wide approach to investigate the changes in acetylation at the transcriptome level in the SDH of rats with CIBP, the primary mechanism of acetylation regulation, and to identify candidates for the treatment of CIBP from the perspective of epigenetic target molecules. We found a wide distribution of ac4C within the rat transcriptome, most sites occurring within CDS, and found that acetylation at the transcriptomic level in the SDH of rats was significantly altered when CIBP occurred. We identified 627 genes in the CIBP model, and their ac4C peak expression increased (Figure 3). Through the verification experiment of screening, the siRNA sequence targeting rat NAT10 in some candidate genes inhibited the expression change of the ac4C peak and inhibited the expression of the target gene, suggesting a regulatory role of acetylation in the expression of target genes by increasing the half-life of mRNA (Figure 8). A sketch showing the hypothesis that bone cancer leads to increased activity of NAT10, an RNA ac4C-modifying enzyme in SDH neurons, promoting RNA acetylation of some CIBP-related genes, improves mRNA stability, prolongs half-life, and promotes the expression of target genes.
In the present study, multiple differential ac4C acetylation genes in CIBP were detected and analyzed, indicating their involvement in several important biological processes, including Nfat5, Pafah1b1, Srgap3, and Nrxn2. These findings offer insights for future research. The pathogenesis and development mechanism of CIBP provides potential targets for treating CIBP. Since strand-specific libraries were constructed and sequenced, mRNAs and lncRNAs containing ac4C peak sites were detected, and their differential ac4C acetylation sites were also identified. LncRNAs play important roles in chromatin remodeling and transcriptional and post-transcriptional regulation. However, the effect of ac4C acetylation on lncRNA function remains unclear. This study is the first to identify differences in the ac4C acetylation modification of lncRNAs in CIBP rats, affecting the role of lncRNAs.
Our study characterized the differential ac4C acetylation in CIBP rats compared to the sham rats, implying a strong correlation between ac4C acetylation and the regulation of CIBP-related gene expression in the SDH of the rat, thus contributing significantly to upcoming research aimed at enhancing CIBP treatment.
Supplemental Material
Supplemental Material - Epitranscriptomic profiling of N4-acetylcytidine-related RNA acetylation in the spinal dorsal horn of rat with cancer-induced bone pain
Supplemental Material for Epitranscriptomic profiling of N4-acetylcytidine-related RNA acetylation in the spinal dorsal horn of rat with cancer-induced bone pain by Longsheng Xu, Shang Zheng, Beibei Liu, Chengfei Xu, Lei Yang, Qinghe Zhou, Ming Yao and Xiang-yao Li in Molecular Pain.
Footnotes
Acknowledgements
We thank Home for Researchers editorial team (www.home-for ![]() ) for language editing service.
) for language editing service.
Author contributions
L.X. and X.L. performed experiments and analyzed data; S.Z., B.L., C.X., and Y.L. interpreted the results of experiments; Q.Z. and L.X. prepared figures; L.X. drafted the manuscript; Q.Z. and X.L. edited and revised the manuscript; X.L. conceived and designed the research. All authors approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported, in part, by grants from the Natural Science Foundation of Zhejiang Province (LTGC23H090002), National Natural Science Foundation of China (82001176), Medical and Health Research Project of Zhejiang Province (2023KY1195), Zhejiang Multidisciplinary Innovation Team of Traditional Chinese Medicine for Diagnosis and Treatment of Elderly Headache and Vertigo (2022-19), Key Discipline Established by Zhejiang Province and Jiaxing City Jointly --Pain Medicine (2019-ss-ttyx) and Jiaxing Key Laboratory of Neurology and Pain Medicine.
Data availability statement
All sequencing data are available from GEO under the accession number GSE225571.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.