
Abstract
Chronic pain is a refractory health disease worldwide causing an enormous economic burden on individuals and society. Accumulating evidence suggests that inflammation in the peripheral nervous system (PNS) and central nervous system (CNS) is the major factor in the pathogenesis of chronic pain. The inflammation in the early- and late phase may have distinctive effects on the initiation and resolution of pain, which can be viewed as friend or foe. On the one hand, painful injuries lead to the activation of glial cells and immune cells in the PNS, releasing pro-inflammatory mediators, which contribute to the sensitization of nociceptors, leading to chronic pain; neuroinflammation in the CNS drives central sensitization and promotes the development of chronic pain. On the other hand, macrophages and glial cells of PNS and CNS promote pain resolution via anti-inflammatory mediators and specialized pro-resolving mediators (SPMs). In this review, we provide an overview of the current understanding of inflammation in the deterioration and resolution of pain. Further, we summarize a number of novel strategies that can be used to prevent and treat chronic pain by controlling inflammation. This comprehensive view of the relationship between inflammation and chronic pain and its specific mechanism will provide novel targets for the treatment of chronic pain.
Keywords
Introduction
Prevalence of chronic pain in different countries and regions.

Pain research in the past decades has confirmed that neuronal plasticity is the critical mechanism for the initiation and maintenance of chronic pain.
18
The neuronal plasticity derives not only from the adaptive transformation of neurons themselves, but also from the change of microenvironment composed of immune and glial cells underlying acute and chronic pain. Under inflammatory conditions, there are increased levels of inflammatory mediators in the tissue from immune and glial cells and even neurons, which modulate nociceptors in the PNS and the pathways responsible for pain transmission in the CNS.
1
Neuroinflammation, a special manifestation of tissue inflammation, can occur in the PNS and CNS, the main characteristics of which are the activation of glial cells and the release of neuroinflammatory mediators (Figure 1).19,20 The cross-talk between neurons and non-neuronal cells leads to deterioration or resolution of pain. Non-neuronal cells [e.g. Schwann cells (SCs), satellite glial cells (SGCs), microglial cells, astrocytes, oligodendrocytes, macrophages, and neutrophils], produce both pro-inflammatory mediators (RED) and anti-inflammatory mediators (BLUE), which can act on their corresponding receptors of the nociceptors causing peripheral and central sensitization marked by a state of hypersensitivity and hyperexcitability of the nociceptor. The central terminals of the nociceptors in the spinal cord form nociceptive synapses with postsynaptic neurons to process pain transmission in the CNS. In addition, descending pathways also exert an inhibitory effect on pain transmission in the spinal cord.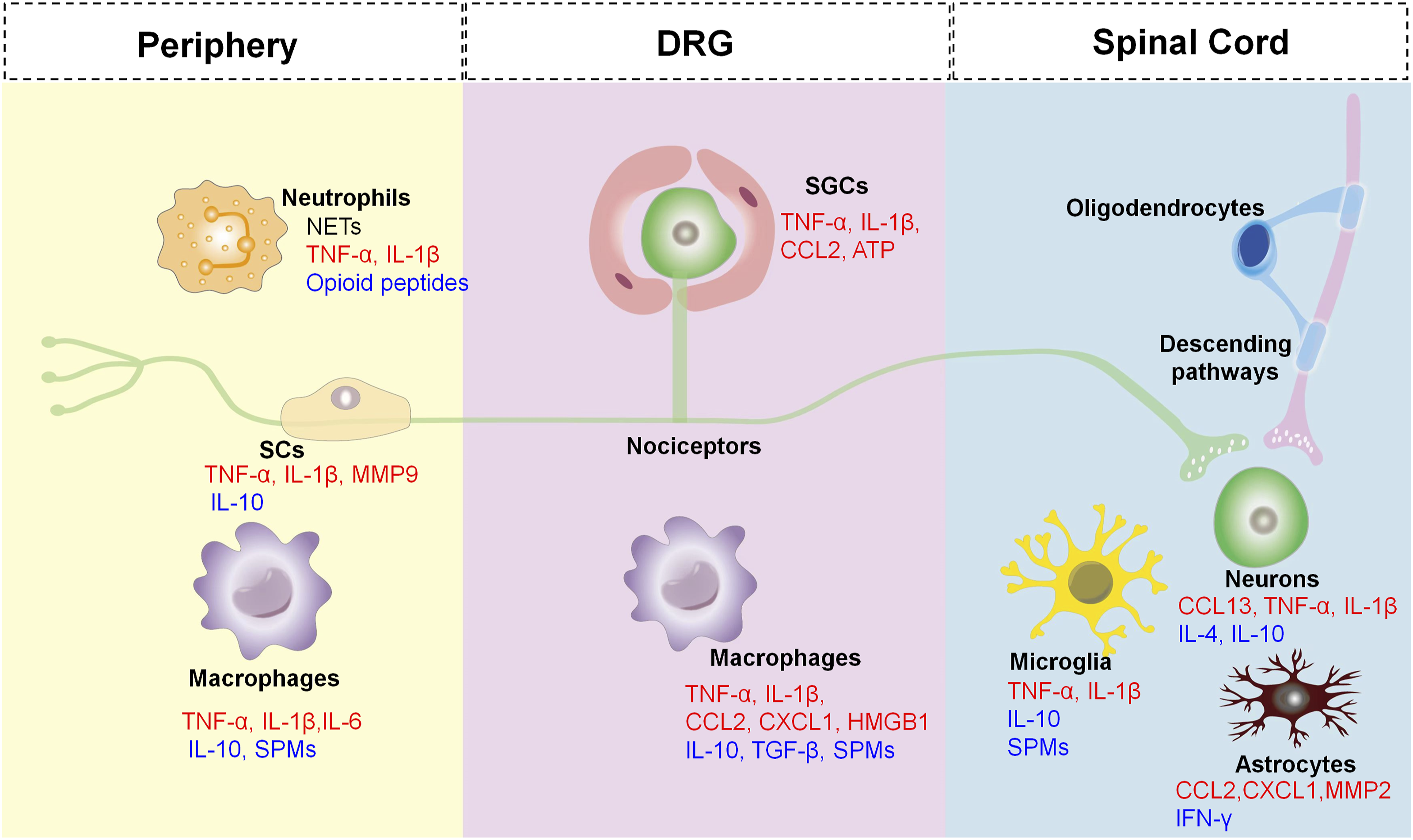
Anti-inflammatory mechanisms of SPMs in chronic pain.

Peripheral inflammation contributes to the pathogenesis of chronic pain
Over the past few decades, accumulating evidence has revealed inflammation in the physiological and pathophysiological processes of chronic pain. Key participants involved are neutrophils, macrophages, and glial cells. In addition, inflammatory mediators (pro-inflammatory cytokines, chemokines), and SPMs also play a significant role in initiation and resolution of chronic pain (Figure 2). The neuroinflammation generated by peripheral glial cells in chronic pain conditions is more persistent, thereby contributing to the maintenance of chronic pain.
50
Inflammation and peripheral sensitization in chronic pain. Inflammation manifests the releasing of inflammatory mediators from various types of cells, these inflammatory mediators including proinflammatory cytokines, chemokines, HMGB1 and ATP, which can bind with their respective receptors on the nociceptive neurons in peripheral, modulate their sensitivity and excitability, and then lead to chronic pain. In addition, SCs and macrophages also produce anti-inflammatory cytokines and pro-resolving mediators SPMs, which promote pain resolution.
Pain modulation by various cells in PNS
An increase in peripheral sensitization is characterized by the hyperexcitability and hypersensitivity of nociceptors caused by tissue injury and inflammation. Increasing evidence suggests that peripheral sensitization is the cardinal contributor to chronic pain in the peripheral, but not an independent factor. There are a variety of non-neuronal cells, such as immune cells and glial cells, are activated along the pain circuitry by painful injuries, causing a localized inflammation in the PNS and CNS (neuroinflammation). The activation of these cells, in turn, causes bi-directional interactions with nociceptors, which are crucial in the development and maintenance of chronic pain. 2 In addition, inflammatory mediators, such as cytokines and chemokines, produced by these cells also contribute to the initiation and maintenance of pain.51,52 Comparatively, SPMs have been proven to promote the resolution of inflammation, alleviate pain, and promote tissue regeneration via specific cellular and molecular mechanisms. Therefore, SPMs exhibit potent anti-inflammatory actions and act as promising analgesics during the resolution phase of inflammation.53–55
Neurons
A variety of noxious/painful stimuli are detected by neurons, which represent a heterogeneous neuron population that can be further classified in accordance with their characteristics, such as size of the soma, degree of myelination, expression of genes, cell surface markers, electrophysiological characteristics.56–58 Afferents that respond to tissue injury include myelinated C-fibers and unmyelinated C-fibers that terminate in skin, muscle, joints, and visceral organs, with their neuronal bodies located in the dorsal root ganglia (DRGs) and trigeminal ganglia (TGs).
These neurons send centrally-projecting fibers to the dorsal horn (for DRG neurons) and spinal trigeminal nuclei of the medulla (for TG neurons), where they convey information to secondary nociceptive neurons via various neurotransmitters and neuropeptides.18,58
Nociceptors are sensitized by inflammatory mediators (e.g. pro-inflammatory cytokines, chemokines, nerve growth factor, bradykinin, prostaglandins)18,59–61 via their corresponding receptors [e.g. G-protein coupled receptors (GPCRs), tyrosine kinase receptors, and ionotropic receptors] that expressed on the terminals and/or cell bodies of nociceptors.18,62 Peripheral receptor activation under pathological conditions cause hyperexcitability and hypersensitivity of nociceptive neurons (peripheral sensitization), via modulation of various ion channels, including transient receptor potential ion channels (TRPs, e.g. TRPA1, TRPV1, and TRPV4),63–65 sodium channels (e.g. Nav1.7/1.8/1.9),66–68 and mechanosensitive piezo ion channels.69,70 The activation of protein kinases [e.g. protein kinase C (PKC) and protein kinase A (PKA), MAP kinases] in primary sensory neurons also participates in peripheral sensitization. For instance, during the pathogenesis of chronic pain, the activation of p38 in sensory neurons initiates and maintains peripheral sensitization by increasing TRPV1 activity in response to tumor necrosis factor-α (TNF-α)71,72 and also by increasing the activity of Nav1.8 as a consequence of interleukin (IL)-1β.73,74 A recent study indicated that TLR8 in TG neurons facilitates trigeminal neuropathic pain (TNP) via increasing intracellular Ca2+, activating ERK and p38, and further increasing the pro-inflammatory cytokines (e.g. TNF-α, IL-6, and IL-1β) in the TG after infraorbital nerve injury. 75 The CXCR3 activation by C-X-C motif chemokine ligand (CXCL) 10 in DRG neurons enhances neuronal excitability via activating p38 and ERK, which contributes to the maintenance of neuropathic pain. 52
A great deal of evidence suggests that peripheral sensory neurons not only respond to painful stimuli, but also play a role in inflammation and immunity as well. 76 Recent research has shown that peripheral sensory neurons express the proton-selective ion channel Hv1 that was previously known to be selectively expressed in microglia. Neuronal Hv1 is upregulated by peripheral inflammation, which in turn enhances inflammation and nociception. Inhibition of neuronal Hv1 by gene knockout or a selective blocker YHV98-4 reduces intracellular ROS production and alkalization in chronic inflammatory pain, restores downstream SHP-1-pAKT signaling, diminishes the release of chemokines, and finally reduces the effects of morphine-induced hyperalgesia and tolerance. 76
Peripheral glial cells: Schwann cells and satellite glial cells
Glial cells in the PNS consist of Schwann cells (SCs) in the peripheral nerve trunk and satellite glial cells (SGCs) in the DRGs and TGs. As a result of noxious stimuli, glial cells release multiple inflammatory mediators, sensitizing nociceptors at axons and somata which in turn participate in pain processing. 2
SCs are the main glial cells of the PNS, which form myelin sheaths to provide nutrients for nerve fibers and maintain the homeostasis of the neuronal microenvironment. 77 After peripheral nerve injury, the activated SCs release pro-inflammatory cytokines, such as TNF-α and IL-1β, which contribute to nerve degeneration and the development of neuropathic pain.78,79 For instance, experimental studies of chronic constriction injury (CCI) rat models have shown that SCs are activated and release the IL-1β at the injury site.80,81 An increase of IL-1β then promotes SCs de-differentiation in Wallerian degeneration via the c-Jun/AP-1 signal axis. 82 Activated SCs can destruct the blood-nerve barrier through the secretion of MMP-9, which recruits immune cells from the blood vessels into the injured nerve tissue and then releases more pro-nociceptive mediators. 83 Consequently, the increased pro-inflammatory mediators and activated SCs are implicated in pathological pain via glial-neuronal and neuro-immune interactions.
SGCs surround the somata of DRG and TG neurons and are coupled with each other through gap junctions, which are activated and proliferated due to injured nerves and inflammation. The activity of SGCs play a key role by participating in chronic pain through the following ways: (1) upregulation of glial fibrillary acidic protein (GFAP),84,85 (2) enhanced gap junction-mediated SGC-SGC and neuron-SGC coupling,86–88 (3) increased the sensitivity of SGCs to ATP that activates P2X7 receptors leading to TNF-α release. By contrast, it enhanced the P2X3 receptor-mediated responses and increased DRG excitability,89–91 (4) decreased the expression of the Kir4.1,92–94 and increased the release of pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, and CX3CL1.95–98 A recent study has shown that the upregulation of P2Y14 receptors in TG SGCs may partially trigger orofacial inflammatory pain via activating ERK and p38 signaling, producing cytokines (IL-1β, TNF-α, and CCL2). 99
Neutrophils and macrophages
Neutrophils are considered as the body’s first line of defense against infection and various inflammatory signals, also viewed as important players in tissue repair. In the early stage of inflammation, neutrophils can fight against infections via phagocytosis and/or neutrophil extracellular traps (NETs). Neutrophils may exert analgesic effects by releasing opioid peptides, such as β-endorphin, met-enkephalin, and dynorphin A. 100 However, when the inflammation persists, neutrophils also release NETs to exacerbate tissue inflammation. 101 Parisien et al. reported that transient neutrophil-induced inflammatory responses are protective against the transition from acute to chronic pain in participants with resolved pain within 3 months. A lack of neutrophils delays the resolution of chronic pain, whereas a supplement of neutrophils prevents the development of prolonged pain. 102 Thus, the inhibition of acute inflammation may delay the resolution of chronic pain. In addition, the enhanced phagocytosis of apoptotic neutrophils by macrophages (also termed efferocytosis) has been revealed to reduce inflammation in synovitis. 103
A neuroimmune crosstalk contributes to chronic pathological pain including inflammatory and neuropathic components.104,105 Macrophages play an important role in pain induction and maintenance with different mechanisms in the periphery. 2 The main functions of these cells in the immune system include phagocytosis, antigen presentation, and cytokine production. It is well known that monocytes differentiate into macrophages in tissues where they fight infection and repair wounds under inflammatory conditions. 106 These cells secret multiple mediators [e.g. TNF-α, IL-6, IL-1β] to regulate the excitability of the primary afferents104,107 via modulation of ion channels (e.g. TRPA1, TRPV1, and Nav1.7–1.9),18,50,53 which then enhance pain transduction and conduction.
In addition to pro-inflammatory cytokines, macrophages also release chemokines which contribute to the occurrence and maintenance of chronic pain. 108 Macrophages can produce chemokines CCL2. 17 In a rat model of lumbar disc herniation, CCL2 was increased in DRG neurons, and its receptor CCR2 was increased in DRG macrophages.109,110 Thus, CCL2/CCR2 signaling may be involved in the interaction of neuron-macrophages and contribute to the maintenance of chronic pain.
In the inflammatory microenvironment of local tissue, the macrophages recruited and resident undergo phenotypic and functional changes responding to mediators released by neurons or immune cells.111–113 Pro-inflammatory macrophages are classified as M1 and anti-inflammatory macrophages are classified as M2. Lipopolysaccharide (LPS), granulocyte/macrophage colony-stimulating factor (GM-CSF), interferon-γ (IFN-γ), and TNF-α polarize macrophage M1 to produce marked effects on inhibiting microbes and tumors, and promoting tissue damage via the production of pro-inflammatory mediators. Transforming growth factor-β (TGF-β), macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, and IL-13 polarize macrophage M2 to phagocytose apoptotic cells and promote tissue repair by secreting anti-proinflammatory mediators IL-10 and TGF-β. 104 After the peripheral nerve and/or adjacent tissues are damaged, macrophages cluster at the injured site, and along primary afferents and its bodies, where M1 macrophages sensitize the nociceptive neurons, which might be subsequently suppressed by M2 macrophages.50,114 M1 macrophages release abundant inflammatory mediators, such as TNF-α, IL-6, IL-1β, nerve growth factor (NGF), insulin-like growth factor 1 (IGF-1), prostaglandin E2 (PGE2), CCL2, and CXCL1, to result in sensitized neurons by activating their specific receptors located in the nociceptive neurons. 104 An increase of high mobility group box 1 (HMGB1) from the activated M1 macrophages stimulates its membrane receptors, such as Toll-like receptor 4 (TLR4), advanced glycation end-product (RAGE), and then accelerates the CXCL12/CXCR4 signal axis in the peripheral nerve, and finally contributes to the pathogenesis of chronic pain.104,115–119
As described above, activated macrophages (mainly M1 macrophages) induce inflammation through releasing pro-inflammatory mediators resulting in neuronal sensitization and then causing pain. Thus, inflammation acts as a role of foe in such situations. On the other hand, M2 macrophages release anti-inflammatory cytokines to suppress inflammation in the resolution phase and resolve pain. As literature reported, the GPR37 in macrophages were activated by neuroprotectin D1 (NPD1) to enhance macrophage phagocytosis and IL-10 production, and then promote the resolution of inflammatory pain. 120 Thus, macrophage activation under these circumstances plays a friend role in pain resolution.
Specialized pro-resolving mediators
SPMs are produced during the resolution phase of inflammation, including various bioactive molecules, such as resolvins (RvD1, RvD2, RvD5, RvE1), 121 maresin (MaR1 and MaR2),55,122,123 protectin (PD1)124,125 or neuroprotectin (NPD1),17,126 and lipoxins,127,128 which derive from polyunsaturated fatty acids (PUFA).44,53–55,129 The synthesis of SPMs plays a crucial role in promoting transformation from a pro-inflammatory to an anti-inflammatory state, boosting a pro-resolution state, and further alleviating inflammatory pain. 123 In addition, SPMs might suppress the excessive sensitization of nociceptors via inhibiting specific channels (e.g. TRPA1 and TRPV1) to achieve the resolution of pain. 126 For example, the peripheral (intraplantar) or spinal (intrathecal) administration of RvE1 and RvD1 has shown a powerful effect on reducing inflammatory pain induced by formalin, carrageenan, or complete Freund’s adjuvant (CFA). Moreover, intrathecal injection of RvE1 inhibits spontaneous pain and heat and mechanical hypersensitivity induced by capsaicin and TNF-α. RvE1 also inhibits TNF-α and TRPV1-induced excitatory postsynaptic current increases via inhibition of ERK signaling pathway. 127 MaR1 and PD1 can suppress the activity of TRPV1 in DRG neurons and reduce neuroinflammatory pain by inhibiting PKA and ERK activity.128,129 MaR2 is a DHA-derived SPM produced by macrophages, which also demonstrates the ability to reduce TRPV1 and TRPA1 activation in DRG neurons exhibited by a reduction in calcium influx and suppressed pain-like behavior in LPS-induced inflammatory pain models, as well as capsaicin and AITC-induced spontaneous pain models of mouse. 44 Moreover, PD1 also decreases hyperexcitability by negatively regulating LTP in the spinal cord evoked by TNF-α. 130 Due to the serious side effects of traditional analgesics, SPMs have become a promising substitute for pain relief, for its potent pro-resolving and anti-inflammatory effects. 123
Central inflammation contributes to the pathogenesis of chronic pain
In the CNS, it is believed that chronic pain is persisted by central sensitization caused by synaptic plasticity in central pain pathways and an increase in neuronal responsiveness as a result of painful insults. Increasing evidence shows that central sensitization is caused by neuroinflammation in the CNS. A classical feature of neuroinflammation is that the glial cells are activated in the CNS, and they release multiple pro-inflammatory mediators to induce hyperalgesia and allodynia (Figure 1).
Pain modulation by central glial cells
Neuroinflammation in the CNS is caused by the activation of glial cells (such as microglia and astrocytes), which play a critical role in the initiation and maintenance of chronic pain.131–135 Tissue inflammation or nerve injury induces the activation of glial cells, which leads to the release of inflammatory mediators, such as cytokines (e.g. TNF-α, IL-6, and IL-1β), chemokines (e.g. CXCL1, CX3CL1, and CCL2), prostaglandin E2 (PGE2), and inducible nitric oxide synthase (iNOS), to enhance neuronal excitability or synaptic transmission.136–138
Microglia are the resident immune cells in CNS. Microglia become activated (also termed “microgliosis”) in response to disease states or peripheral nerve injury, characterized by significant morphological changes (hypertrophy), functional changes, and proliferation, which is related to transcriptomic changes. 139 The spinal microglia activation is evoked by pro-inflammatory mediators [e.g. IL-1β, caspase-6, colony-stimulating factor 1 (CSF1), and extracellular proteases] derived from sensory neurons.139–143 After nerve injury, activation of microglia in the ipsilateral spinal dorsal horn can be quickly observed, and inhibition of microglia activation is sufficient to mitigate pain development. 144 However, it should be noted that microgliosis induced by nerve injury is transient and self-limiting, which weakens within a few weeks and remains at a reduced level in the later stage, that means in the later stage of microgliosis, microglia inhibitor and inhibition of activated microglia-associated cytokines are no longer effective in pain relief.145,146 Thus, microglia and their pain mediators (cytokines and chemokines) are most actively involved in pain initiation. Of note, SPMs in CNS have been shown to contribute to the resolution of pain. The NPD1 biosynthesized by microglia has a powerful protective effect in pain relief. 147 In addition, microglia express many SPM receptors, such as GPR18 (RvD2 receptor), GPR32 (RvD1 receptor), and ChemR23 (RvE1 and RvE2 receptor).148,149 RvE1 binds to ChemR23 to trigger pro-resolving responses and reduce chronic pain through inhibiting microglial activation in the SC.150,151 Consequently, activation of these microglial receptors down-regulates the production of pro-inflammatory mediators (e.g. TNF-α, IL-1β), and up-regulates the secretion of anti-inflammatory mediators (e.g. IL-10, TGF-β), thus promoting the regression of pathological pain. 139 Besides, ion channels on microglia are involved in regulation of inflammatory pain, such as Hv1, P2X4, P2X7, and TRPV4. Hv1 has been implicated in the pathogenesis of neuropathic pain by regulating ROS production and microglia-astrocyte communication. 152 The microglial P2X4 channel participates in inflammatory pain through activating microglia, and then releasing pro-inflammatory mediators. 153 The ATP-gated ion channel P2X7 on microglia have been revealed to modulate inflammatory pain through regulating NLRP3/Caspase-1 pathway. 154 TRPV4 expression in spinal resident microglia is increased in the mouse model of SNI, which transforms peripheral nerve injury into central sensitization and neuropathic pain through releasing lipocalin-2. 155
Astrocytes are more abundant (approximately 2-4 times) compared to microglia, and play crucial effects on controlling the extracellular microenvironment, repairing the imbalance of potassium, glutamate, and water homeostasis under normal physiological conditions.156,157 Astrocytes are activated in various pathological conditions and convert to reactive states (also termed “astrogliosis”) characterized by obvious morphological changes, a significant increase of GFAP, and a significant cellular proliferation, which is considered to be coupled with a loss of homeostatic functions of astrocytes.58,158 Astrocytes are activated after nerve injury generally following microglial activation (e.g. 1 week), but remain longer time (at least 5 months or more) in a reactive state that contributes to the maintenance of chronic pain. 159 Numerous pieces of evidence show that decreasing activation of astrocyte can attenuate pain behaviors in chronic pain models.160–162
Glia in the CNS are dominated by oligodendrocytes, which account for 46∼75%. 58 There is, however, relatively little research on the role of oligodendrocytes in chronic pain pathogenesis. Some studies suggest that there may be an interaction between oligodendrocyte damage and pain in patients with multiple sclerosis. 163 Moreover, oligodendrocytes play a role in maintaining pain in common conditions. 164 Previous experiments suggest that the role of oligodendrocytes in pain regulation may be connected with primary afferent nerve, microglia, and astrocytes. 165 Oligodendrocyte-derived IL-33 in the dorsal SC plays an important role in neuropathic pain. IL-33 is increased in neuropathic pain model, and its receptor IL-33R (ST2) deficiency has been exhibited analgesic effects. IL-33-induced pain has been significantly alleviated through inhibition of PI3K, MAPKs (p38, ERK, and JNK), and NF-κB. 165
Following peripheral nerve injury (e.g. SNI), microglia are generally thought to activate before astrocytes. Microglia activation in the early stage is necessary for the initial generation of acute pain. Activated microglia secret pro-inflammatory mediators to increase sensitivity of nearby nociceptive neurons and astrocytes, resulting in astrogliosis, which contributes to the maintenance of chronic pain. 58 Understanding the role of inflammation in different stages is helpful for intervention selections for pain relief.
Neurons in the spinal cord
Chemokines combined with their receptors in spinal neurons contribute to the establishment and maintenance of chronic pain. For instance, CXCL1 and CCL2, which are expressed in spinal astrocytes, act respectively on CXCR2 and CCR2 in spinal neurons after spinal nerve ligation (SNL) to enhance excitatory synaptic transmission (astrocyte-to-neuron signal axis). CXCL13 in spinal neurons is also highly up-regulated after SNL and induces spinal astrocyte activation through its receptor CXCR5 (neuron-to-astrocyte signal axis). 166
Descending pathways have been revealed to modulate pain transmission in spinal cord, such as the anterior cingulate cortex (ACC)-spinal cord pathway, noradrenergic descending pathways, and dopaminergic descending pathways.167–169 Descending pathway of the ACC-spinal cord facilitates SNI-induced neuropathic pain through a rostral ventromedial medulla (RVM)-independent manner. 167 Descending noradrenergic pathways also exert an inhibitory effect on pain transmission in chronic pain models.170–172 Peripheral inflammation activates the descending noradrenergic pathways from the locus coeruleus (LC) to the spinal cord, which prevents the hyperalgesia development. 173 As a result of unilateral hind paw inflammation, descending NE-containing neurons in the LC are stimulated, increasing the level of NE in the ipsilateral dorsal horn. 174 A recent study has reported that activation of LC-spinal cord noradrenergic neurons alleviated neuropathic pain in mice via down-regulating the expression of pro-inflammatory cytokines (TNF-α and IL-1β), up-regulating the expression of anti-inflammatory cytokines (IL-4 and IL-10), and inhibiting activation of microglia and astrocytes in SDH. 175
Conclusion and prospects
Given the important role of inflammation (including neuroinflammation) in the induction and maintenance of chronic pain, it may be effective to directly target inflammation through cytokines, chemokines, and MAP kinase inhibitors. There are also possible side effects of these drugs, such as infection after long-term use and an inability to resolve inflammation. Thus, some alternative approaches that can control an excess of inflammation are emerging, including SPMs and cell therapies.
A large number of research suggest that microglia play a vital role in pain generation. When microglia are activated, pro-inflammatory and anti-inflammatory factors are produced in an imbalance. Therefore, discovering original ways to stimulate endogenous anti-inflammatory factors and inhibit pro-inflammatory factors may still be the challenge for researchers. It is generally known that increased production of pro-inflammatory cytokines (e.g. TNFα, IL-6, and IL-1β) mediates allodynia and hyperalgesia. 176 Thus, inhibiting microglial activation by decreasing the production of pro-inflammatory cytokines may have an analgesic effect. TNF-α blocker etanercept has been reported to reduce neuropathic pain in a rodent model of spinal cord injury through intrathecal administration, 177 and administration of etanercept to patients with severe sciatica also showed effective. 178 Administration of anti-IL-1β antibodies also demonstrated a reduce of neuropathic pain. 179
The chemokines not only regulate inflammation in the immune system, but also causes neuroinflammation in PNS and CNS, and promotes the initiation and maintenance of various types of chronic pain. Thus, the chemokine system provides an ideal candidate for treatments of chronic pain. According to the preclinical studies, there are three candidate directions for the therapeutic drugs targeting chemokine and its receptor: (1) blocking or neutralizing antibodies; (2) small-molecule inhibitors; (3) therapeutic small interfering RNA (siRNA). The antibodies of CGRP and its receptors have been used to treat migraine by subcutaneous or intravenous injection. 180 The small molecule CCR5 antagonist maraviroc has been observed to alleviate neuropathic pain in clinical practice.181,182
Many studies have proved that SPMs can control inflammation effectively and hold the potential to suppress pain while promoting pain relief. However, the instability and high production cost of SPMs limits their direct application to patients. 183 Omega-3 fatty acids (such as DHA and EPA) derived from food are precursors of SPMs and have been shown to reduce inflammatory pain in patients, which may be through its own GPCRs, such as GRP120.184,185 A randomized trial has shown that dietary omega-3 fatty acids may alleviate headaches in patients. 186 However, the efficacy of these SPMs precursors for pain relief is much lower compared to their metabolites. Thus, it will be promising to study the synergistic effects of SPM precursors from diet and neuromodulation on chronic pain will be promising, since it targets inflammation and neuroinflammation.
Currently, there has been a rise in gene therapy that is practiced in various inherited diseases in clinics. The recombinant adeno-associated virus (rAAV) is often used as a vector to deliver genes in gene therapy because the rAAV lacks any detrimental viral gene, does not integrate into the genome of humans, and provides persistent gene expression. With respect to the benefits of rAAV, gene therapy will be a promising treatment to tackle the ticklish chronic pain by silencing and increasing the pro-inflammatory and anti-inflammatory mediators, respectively.
Footnotes
Author contributions
Xiao-Xia Fang wrote the manuscript. Meng-Nan Zhai designed the tables. Meixuan Zhu participated in writing and editing the manuscript. Cheng He, Heng Wang, and Juan Wang drew the figures. Zhi-Jun Zhang initiated and supervised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (NSFC) (31970938, 81571070) and the Natural Science Research Program of Jiangsu Province (BK20191448).