
Abstract
Background
Chemokine-mediated neuroinflammation plays an important role in the pathogenesis of neuropathic pain. The chemokine CC motif ligand 7 (CCL7) and its receptor CCR2 have been reported to contribute to neuropathic pain via astrocyte-microglial interaction in the spinal cord. Whether CCL7 in the trigeminal ganglion (TG) involves in trigeminal neuropathic pain and the involved mechanism remain largely unknown.
Methods
The partial infraorbital nerve transection (pIONT) was used to induce trigeminal neuropathic pain in mice. The expression of Ccl7, Ccr1, Ccr2, and Ccr3 was examined by real-time quantitative polymerase chain reaction. The distribution of CCL7, CCR2, and CCR3 was detected by immunofluorescence double-staining. The activation of extracellular signal-regulated kinase (ERK) was examined by Western blot and immunofluorescence. The effect of CCL7 on neuronal excitability was tested by whole-cell patch clamp recording. The effect of selective antagonists for CCR1, CCR2, and CCR3 on pain hypersensitivity was checked by behavioral testing.
Results
Ccl7 was persistently increased in neurons of TG after pIONT, and specific inhibition of CCL7 in the TG effectively relieved pIONT-induced orofacial mechanical allodynia. Intra-TG injection of recombinant CCL7 induced mechanical allodynia and increased the phosphorylation of ERK in the TG. Incubation of CCL7 with TG neurons also dose-dependently enhanced the neuronal excitability. Furthermore, pIONT increased the expression of CCL7 receptors Ccr1, Ccr2, and Ccr3. The intra-TG injection of the specific antagonist of CCR2 or CCR3 but not of CCR1 alleviated pIONT-induced orofacial mechanical allodynia and reduced ERK activation. Immunostaining showed that CCR2 and CCR3 are expressed in TG neurons, and CCL7-induced hyperexcitability of TG neurons was decreased by antagonists of CCR2 or CCR3.
Conclusion
CCL7 activates ERK in TG neurons via CCR2 and CCR3 to enhance neuronal excitability, which contributes to the maintenance of trigeminal neuropathic pain. CCL7-CCR2/CCR3-ERK pathway may be potential targets for treating trigeminal neuropathic pain.
Keywords
Introduction
Trigeminal nerve damage-induced neuropathic pain is a severely debilitating chronic orofacial pain syndrome and often refractory to treatment. However, the pathological mechanisms of this neuropathic pain are still poorly understood. 1 Neuroinflammation mediated by chemokines and cytokines plays an important role in the development and maintenance of chronic pain. 2 Chemokines including CCL2, CCL7, CXCL10, CXCL12, and CXCL13 and their corresponding receptors are up-regulated in the dorsal root ganglion (DRG) and spinal cord after nerve injury, and inhibition of their expression relieves neuropathic pain.3–8 Moreover, CCL2, CXCL13, and CXCL10 are also increased in the trigeminal ganglion (TG) after trigeminal nerve injury and contribute to trigeminal neuropathic pain.9–11
CCL7, also known as monocyte chemoattractant protein (MCP)-3, is a member of CC chemokine subfamily and plays pivotal roles in numerous inflammatory diseases.12–14 It was first identified from the supernatant of cytokine-stimulated human osteosarcoma cells. 15 CCL7 exists in a variety of cell types and involves in innate and adaptive immunity and cardiovascular, diabetes, and kidney diseases. 14 It can be up-regulated by cytokines such as interleukin (IL)-1, tumor necrosis factor (TNF)-α and down-regulated by IL-4 and IL-10.16–22 In the rapidly growing transcriptome database, it showed that chronic constriction injury (CCI) of sciatic nerve increased CCL7 expression in the DRG, spinal cord, and anterior cingulate cortex of rats.4,23 Moreover, CCL7 is highly up-regulated in spinal astrocytes after partial sciatic nerve ligation (pSNL). 5 Our gene microarray data showed that partial infraorbital nerve transection (pIONT) induces CCL7 up-regulation in the TG of mice. 24 However, the function of CCL7 in the TG in orofacial neuropathic pain has not been studied yet.
Many CC chemokines have more than one receptor. 25 CCL7 binds to several receptors including CCR1, CCR2, CCR3, and CCR5.14,26 Imai et al. 5 investigated the role of CCL7/CCR2 in neuropathic pain by pSNL model. CCL7 is produced by spinal astrocytes and acts on CCR2 in microglia, thereby inducing spinal microglial activation. CCL7 can also activate CCR1 27 and CCR3. 28 However, CCL7 binds CCR5 with high affinity without eliciting a functional response and therefore is considered a natural antagonist of CCR5. 29
The binding of chemokine and receptor leads to the activation of intracellular signaling pathways, such as the mitogen-activated protein kinases (MAPKs), phospholipase C (PLC) pathway, and phosphoinositide 3-kinases (PI3K).30,31 The MAPKs members including extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) are activated in the DRG and spinal cord by peripheral nerve injury. 32 Similarly, phosphorylated ERK, p38 and JNK are accumulated, increasing excitability of sensory neurons and thus facilitating mechanical allodynia in trigeminal neuropathic pain models.10,11,24,33,34 Activation of MAPKs leads to the expression of new proteins (e.g. pro-inflammatory mediators) or enhancing the function of key ion channels or receptors (e.g. sodium channels and TPRV1). 35 Intra-ganglionic injection of a p38 selective inhibitor SB203580 or ERK inhibitor PD98059 attenuated partial infraorbital nerve ligation (pIONL)-induced pain and the expression of TNF-α and IL-1β in the TG.10,34 Up to now, whether ERK mediates the function of CCL7 has not been investigated.
Here, we investigated the function and mechanism of CCL7 in regulating neuronal excitability and trigeminal neuropathic pain using pIONT model in mice, hypothesizing that downstream receptors are activated by CCL7 in the TG, which further enhances ERK-dependent neuronal excitability and exacerbates trigeminal neuropathic pain.
Materials and methods
Animals and surgery
ICR mice (male, 6–8 weeks old) were purchased from the Experimental Animal Center of Nantong University. All mice were housed in standard polycarbonate cages under controlled ambient temperature (22–24°C) and humidity range of 40–60%, with a 12:12 h dark-light cycle, and allowed ad libitum access to water and food. The experimental and surgical procedures were reviewed and approved by the Animal Care and Use Committee of Nantong University. Animal treatments were performed in accordance with the guidelines of the International Association for the Study of Pain. For the pIONT surgery, the mouse was anesthetized and laid on the back. The oral cavity was exposed. A 1-mm longitudinal incision on the left buccal mucosa and at the level of the maxillary first molar was made to expose the infraorbital nerve (ION). The ION was then isolated and approximately one half of the nerve was tightly ligated with 6-0 silk suture and then transected just distal to the ligature. The buccal mucosa tissue was then sutured. 33 For the sham-operated mice, the ION was exposed but not ligated or transected.
Drugs and administration
The recombinant mouse CCL7 was purchased from Sino Biological (Beijing, China). Ccl7 siRNA and negative control siRNA (NC siRNA) were designed by Gene Pharma (Suzhou, China). The selective CCR1 antagonist BX471, CCR2 antagonist RS504393, and CCR3 antagonist SB328437 were purchased from Tocris (Bristol, UK). For Intra-TG injection, the animals were anesthetized with isoflurane and drugs were injected with a 30 G needle from the infraorbital foramen to the foramen rotundum. The tip of the needle terminated at the medial part of the TG, and the siRNA or antagonists (5 μL) were slowly delivered.
Real-time quantitative polymerase chain reaction (qPCR)
The primer sequences of genes for qPCR.

Immunohistochemistry
For immunofluorescence staining, mice were deeply anesthetized and transcardially perfused with 4% paraformaldehyde. TG sections were cut at 15 μm on a cryostat and processed for immunostaining as previously described. 37 The following primary antibodies were used: ATF3 (rabbit, 1:1000, Santa Cruz), CCL7 (rabbit, 1:200, Biorbyt), CCR2 (rabbit, 1:50, Bioss), CCR3 (rabbit, 1:50, Boster), TUJ1 (mouse, 1:500, R&D Systems), IBA-1 (goat, 1:500, Abcam), glutamine synthetase (GS, mouse, 1:1000, Millipore), NF200 (mouse, 1:500, Sigma), CGRP (goat, 1:3000, Millipore), IB4-FITC (1:200, Sigma), pERK (rabbit, 1:400, CST). The sections were then incubated with Cy3-or Alexa 488-conjugated secondary antibodies (1:1000, Jackson, West Grove, PA, USA). A Nikon Electron Microscope (Nikon Eclipse NiE, Japan) was used to examine the stained sections and capture images.
Western blot
The TG tissues were homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma). The BCA Protein Assay was used to determine the protein concentrations. SDS-PAGE and Western blot were performed as previously described. 34 The following antibodies were used: pERK (rabbit, 1:1000, CST), ERK (rabbit, 1:1000, CST), and IRDye 800CW donkey anti-rabbit secondary antibody. The images were captured by the Odyssey Imaging System (LI-COR Bioscience). Specific bands were evaluated by apparent molecular size. The intensity of the selected bands was analyzed using Image J software (NIH, Bethesda, MD).
Behavioral testing
Before assessing the facial pain baseline, the mice were habituated to the behavioral test cage for 30 min every day for more than 2 days in the behavioral test environment. As previously described,38,39 two von Frey filaments (0.02 g and 0.16 g) were used to stimulate the ipsilateral infraorbital nerve region, and the response of the mice was recorded. The filaments were applied to stimulate for 3 times. The final nocifensive behavior score of three measurements was calculated according to the following criteria: score 0, no response or looked around; score 1, exploratory behavior - the mouse detected the von Frey filament; score 2, slight withdrawal response—the mouse slowly retracted its face from the stimulation; score 3, quick and intense withdrawal response with paw lifting; score 4, the mouse wiped its face with the forepaw toward the stimulated facial area less than 3 times; score 5, the mouse wiped its face with the forepaw toward the stimulated facial area more than 3 times.
TG neurons preparation and electrophysiological recording
ICR mice (6–8 weeks) were decapitated after being anesthetized with isoflurane, and then the TGs were quickly removed and placed in ice-cold 95% O2 and 5% CO2-saturated artificial cerebrospinal fluid (ACSF) containing the following (in mM): 125 NaCl, 3 KCl, 2.4 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 Glucose, 5 HEPES, adjusted with NaOH to pH 7.4. Under the microscope, the connective tissues of the TG were carefully stripped and then the ganglia were transferred to 1 mL of enzyme solution containing collagenase type I (3.54 mg/mL Gibco, ThermoFisher) and Dispase II (1.65 mg/mL; Roche) for 30 min at 37°C, shaking every 5 min. The ganglia were taken from the enzyme solution, washed and mechanically dissociated with a flame-polished Pasteur pipette. Cells were plated on glass cover slips coated with 0.5 mg/mL poly-d-lysine (Gibco, ThermoFisher) and cultured in a Neurobasal medium (Gibco, ThermoFisher) supplemented with 10% FBS, 2% B27 supplement (Gibco, ThermoFisher), and 1% penicillin–streptomycin (Gibco, ThermoFisher) at 37°C in 5% CO2 for 8–12 h before recording.
The patch-clamp recording experiments were performed at room temperature, and the ganglia were continuously perfused with ACSF saturated with 95% O2 and 5% CO2, where TG neurons could be identified using a ×40 water-immersion objective on a microscope (BX51WI; Olympus). The patch pipettes were pulled from borosilicate glass capillary using a flaming micropipette puller (P-97; Sutter Instruments), and had initial resistance 4∼8 MΩ when filled with the internal pipette solution. Membrane voltage and current were amplified with a Multiclamp 700B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 10 kHz using a data acquisition interface (1440A, Molecular Devices). The pClamp10 software (Axon Instruments) was used for signal acquisition and analysis.
Small diameter TG neurons (<25 μm) were chosen for recording. 40 For current-clamp, the pipette solution containing the following (in mM): 121 potassium gluconate, 20 KCl, 10 Hepes, 0.2 EGTA, 2 MgCl2, 0.4 GTP-Tris, 4 Na2ATP, which was adjusted with KOH to pH 7.4. To study neuronal excitability, action potentials (APs) evoked by a series of square current stimulation (1000 ms in duration and 10 pA increments) were recorded.
Quantification and statistics
All of the results are presented as mean ± SEM. The behavioral data were analyzed by two-way repeated measures (RM) ANOVA followed by the Bonferroni’s test. The qPCR data were analyzed by one-way ANOVA followed by the Bonferroni’s test. For Western blot, the density of specific bands was measured with ImageJ (NIH, USA). Differences between two groups were compared using Student’s t-test. GraphPad Prism v8.0 was used for statistical analyses. p < .05 was considered to be significant (*p < .05, **p < .01, ***p < .001).
Results
pIONT increases CCL7 expression in TG neurons
As previously reported,
24
pIONT induced persistent mechanical allodynia, which started from day 3 and lasted for more than 21 days after the operation (Figure 1(a)). In addition, compared to sham-operated mice, pIONT dramatically increased ATF3-immunoreactivity (IR) neurons in the TG (Figures 1(b) and (c)). We then checked the expression of Ccl7 mRNA in the TG after pIONT or sham-operation. The qPCR showed that Ccl7 mRNA was increased on days 1, 3, 10, and 21 after pIONT (Figure 1(d)). Furthermore, immunofluorescence staining showed that CCL7 has low expression in sham-treated mice and was increased in pIONT-operated mice (Figures 1(e) and (f)). To characterize the cellular localization of CCL7 in the TG, we performed double-staining of CCL7 with the neuronal marker TUJ1, the satellite cell marker GS, and the macrophage marker IBA-1. CCL7 is largely co-localized with TUJ1, but not with GS or IBA-1 (Figures 1(g)–(j)). Furthermore, the double-staining of CCL7 with NF200 (a myelinated neuronal marker), IB4 (a nonpeptidergic nociceptor marker), and CGRP (a peptidergic nociceptor marker) showed that CCL7 is distributed in all neuron types and mainly co-localized with NF200 (Figures 1(k)–(n)). pIONT increases CCL7 expression in TG neurons. (a) pIONT induced mechanical allodynia which started from 3 days and maintained for more than 21 days *p < .05, ***p < .001. Two-way RM ANOVA followed by the Bonferroni’s test. (b) The ATF3 was rarely expressed in sham-operated mice but was increased after pIONT. (c) The percentage of ATF3+ neurons in sham and pIONT groups. ***p < .001, student’s t-test. (d) The Ccl7 mRNA expression in the TG from naive, sham-, and pIONT-operated mice. ***p < .001 vs. corresponding sham group. Student’s t-test. n = 7–8 mice per group. (e) Representative images of CCL7 immunofluorescence in the TG from sham and pIONT mice. CCL7 had low expression in sham-operated mice and was increased in pIONT-operated mice. (f) The percentage of CCL7+ neurons in sham and pIONT groups. ***p < .001, student’s t-test. (g–i) Double immunofluorescence staining shows that CCL7 was mainly colocalized with the neuronal marker TUJ1 (g), but not with the satellite marker GS (h), or the macrophage marker IBA-1 (i) in the TG 3 days after pIONT. (j) The percentage of CCL7/TuJ1 double-stained neurons in CCL7+ neurons. (k–m) Immunofluorescent images show the colocalization of CCL7 with NF200 (k), IB4 (l), and CGRP (m) in the TG 3 days after pIONT. (n) The percentage of CCL7/NF200, CCL7/IB4, and CCL7/CGRP in CCL7+ neurons.
CCL7 contributes to pIONT-induced mechanical allodynia
To determine whether CCL7 plays a role in the development of pIONT-induced trigeminal neuropathic pain, we intra-TG injected Ccl7 siRNA in the TG soon after pIONT and tested pain behaviors 24 h after injection. Behavioral data showed that Ccl7 siRNA decreased the nocifensive score 48 h and 72 h after pIONT, compared with NC siRNA-injected mice (Figure 2(a)). To confirm the knock-down effect of Ccl7 siRNA, we checked mRNA level in another sets of animals 48 h after pIONT. The qPCR showed that Ccl7 siRNA decreased the Ccl7 mRNA level in the TG (Figure 2(b)). To check whether CCL7 is involved in the maintenance of pIONT-induced mechanical allodynia, we injected Ccl7 siRNA 7 days after pIONT. Ccl7 siRNA attenuated mechanical allodynia (Figure 2(c)) at 48 h and also reduced the expression of Ccl7 mRNA (Figure 2(d)). CCL7 contributes to pIONT-induced mechanical allodynia. (a) Intra-TG injection of Ccl7 siRNA after pIONT attenuated pIONT-induced mechanical allodynia. *p < .05, **p < .01, two-way repeated-measures ANOVA followed by the Bonferroni’s tests. The arrow indicates the time of pIONT and siRNA injection. (b) The qPCR showed that Ccl7 siRNA decreased the Ccl7 mRNA level in the TG 48 h after injection. *p < .05, student’s t-test, n = 6 mice/group. (c–d) Intra-TG injection of Ccl7 siRNA 7 days after pIONT attenuated mechanical allodynia (c), and also reduced Ccl7 mRNA expression in the TG (D). *p < .05, n = 5–6 mice/group. The arrow in C indicates the time of siRNA injection. (e) Intra-TG injection of recombinant CCL7 induced facial mechanical allodynia in a dose-dependent manner, **p < .01, ***p < .001, two-way RM ANOVA followed by Bonferroni’s test, n = 8 mice in each group. The arrow indicates the time of CCL7 injection. (f) Intra-TG injection of CCL7 (10 ng) increased pERK level in the TG. ***p < .001, student’s t-test, n = 4 mice/group. (g-i) The pERK distribution in the TG after injection of PBS (g) or CCL7 (10 ng, h). CCL7 induced marked ERK activation in the TG (i). **p < .01, student’s t-test, n = 3 mice/group.
To investigate whether CCL7 is sufficient to induce mechanical allodynia, we injected recombinant CCL7 into the TG of naive mice at a series of doses (1, 10, 100 ng). As shown in Figure 2(e), CCL7 at 10 ng and 100 ng significantly increased the nocifensive score 3 h and 6 h after injection. As ERK plays an important role in trigeminal neuropathic pain, 24 we checked whether CCL7 can activate ERK in the TG. Western blot showed that, compared to PBS injection, CCL7 increased pERK level 3 h after injection (Figure 2(f)). Immunostaining also showed that pERK had low expression in PBS group, but was increased after CCL7 injection (Figures 2(g)–(i)).
CCL7 increases the excitability of TG neurons
The excitability of nociceptive neurons is closely related to neuropathic pain.40,41 To investigate whether CCL7 affects TG neuronal excitability, we performed whole-cell membrane clamp recordings after treatment with CCL7 on small-diameter nociceptive TG neurons. Incubation the neurons with CCL7 (10 ng/mL, 100 ng/mL) for 30 min decreased the rheobase of TG neurons compared with the PBS group (Figures 3(a) and (b)). In addition, CCL7 increased the numbers of APs in response to 160 pA, 1000 ms current injection (Figures 3(c) and (d)). We also examined the effect of CCL7 on the intrinsic membrane properties of TG neurons by analyzing the first AP evoked by step depolarizing current stimulation. Compared with the vehicle, the resting membrane potential (RMP) and APs maximum rise slope (S-Rise) of TG neurons were significantly increased after CCL7 treatment, and the APs threshold and medium AHP amplitude were significantly decreased after CCL7 treatment (Figures 3(e)–(h)). APs amplitude, half-width, maximum decay slope (s-decay), and fast-AHP parameters were not significantly changed (Figures 3(i)–(l)). Collectively, these results suggest that CCL7 enhances the excitability of nociceptive neurons in the TG. CCL7 increases the excitability of TG neurons. (a) The representative traces of APs evoked by a rheobase current from vehicle and CCL7 (10 ng/mL, 100 ng/mL)-treated TG neurons. (b) CCL7 reduced the rheobase current required to evoke an AP. ***p < .001 vs. vehicle, one-way ANOVA followed by the Bonferroni’s tests. n = 20–25 neurons/group from 3–4 mice. (c) The representative traces of APs evoked by depolarizing current steps recorded from vehicle and CCL7 (10 ng/mL, 100 ng/mL)-treated TG neurons. (d) CCL7 (10 ng/ml, 100 ng/mL) increased the number of APs in response to 160 pA, 1000 ms current injection. ***p < .001 vs. vehicle. Two-way RM ANOVA followed by Bonferroni’s tests. (e) CCL7 (100 ng/mL) reduced the RMP of TG neurons. **p < .01 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (f) CCL7 (10 ng/mL, 100 ng/mL) increased the APs s-rise of TG neurons. *p < .05, **p < .01 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (g) CCL7 (10 ng/mL, 100 ng/mL) decreased the APs threshold of TG neurons. *p < .01 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (h) CCL7 (100 ng/mL) decreased the APs medium AHP of TG neurons. *p < .05 vs. vehicle. One-way ANOVA followed Bonferroni’s tests. (i–l) No difference in the APs amplitude (i), half-width (j), s-Decay (k) and fast AHP (l) between vehicle- and CCL7 (10 ng/mL, 100 ng/mL)-treated TG neurons. One-way ANOVA followed by the Bonferroni’s tests.
pIONT increases the expression of CCR1, CCR2, and CCR3
CC chemokine receptor CCR1, CCR2, CCR3, and CCR5 are known as the functional receptors of CCL7.
26
As it was reported that the binding of CCL7 with CCR5 did not elicit a functional response,
29
we examined the mRNA levels of Ccr1, Ccr2, and Ccr3 in the TG after pIONT. The qPCR showed that Ccr1, Ccr2, and Ccr3 were all increased from days 1–21 after pIONT (Figures 4(a)–(c)). We then intra-TG injected BX471 (selective CCR1 antagonist), RS504393 (selective CCR2 antagonist), or SB328437 (selective CCR3 antagonist) 7 days after pIONT to test the analgesic effect. As shown in Figure 4(d), these drugs at the dose of 2 nmol did not affect pIONT-induced mechanical allodynia. However, intra-TG injection of 20 nmol of RS504393 significantly decreased the nocifensive score started from 1 h, maintained at 3 h, and recovered at 6 h (Figure 4(e)). Similarly, intra-TG injection of 20 nmol of SB328437 also relieved mechanical allodynia, which showed effect at 3 h and 6 h. However, the same dose of BX471 had no effect on nocifensive behavior score. We then checked pERK expression in the TG after intra-TG injection of these antagonists (20 nmol) by Western blot. As shown in Figures 4(f)–(h), compared to vehicle injection, intra-TG injection of RS504393 or SB328437, but not BX471 decreased pERK level. These data suggest that CCL7 mediates the activation of ERK signaling via CCR2 and CCR3, thus causing mechanical allodynia. pIONT increases the expression of CCR1, CCR2, and CCR3. (a–c) Ccr1, Ccr2, and Ccr3 mRNA expression in the TG in naive, sham-, and pIONT-operated mice. *p < .05, **p < .01, ***p < .001 vs. corresponding sham group. Student’s t-test. n = 6–8 mice per group. (d) Intra-TG injection of BX471, RS504393, or SB328437 at the dose of 2 nmol had no effect on pIONT-induced mechanical allodynia. (e) Intra-TG injection of RS504393 or SB328437 at the dose of 20 nmol significantly decreased the nocifensive score. *p < .05, ***p < .001. Two-way RM ANOVA followed by the Bonferroni’s test. n = 6–8 mice in each group. The arrows in D and E indicate the time of drug injection. (f–h) Intra-TG injection of RS504393 or SB328437 decreased pERK expression in the TG after pIONT, whereas BX471 had no effect. ***p < .001, student’s t-test. n.s., not significant. n = 4 mice/group.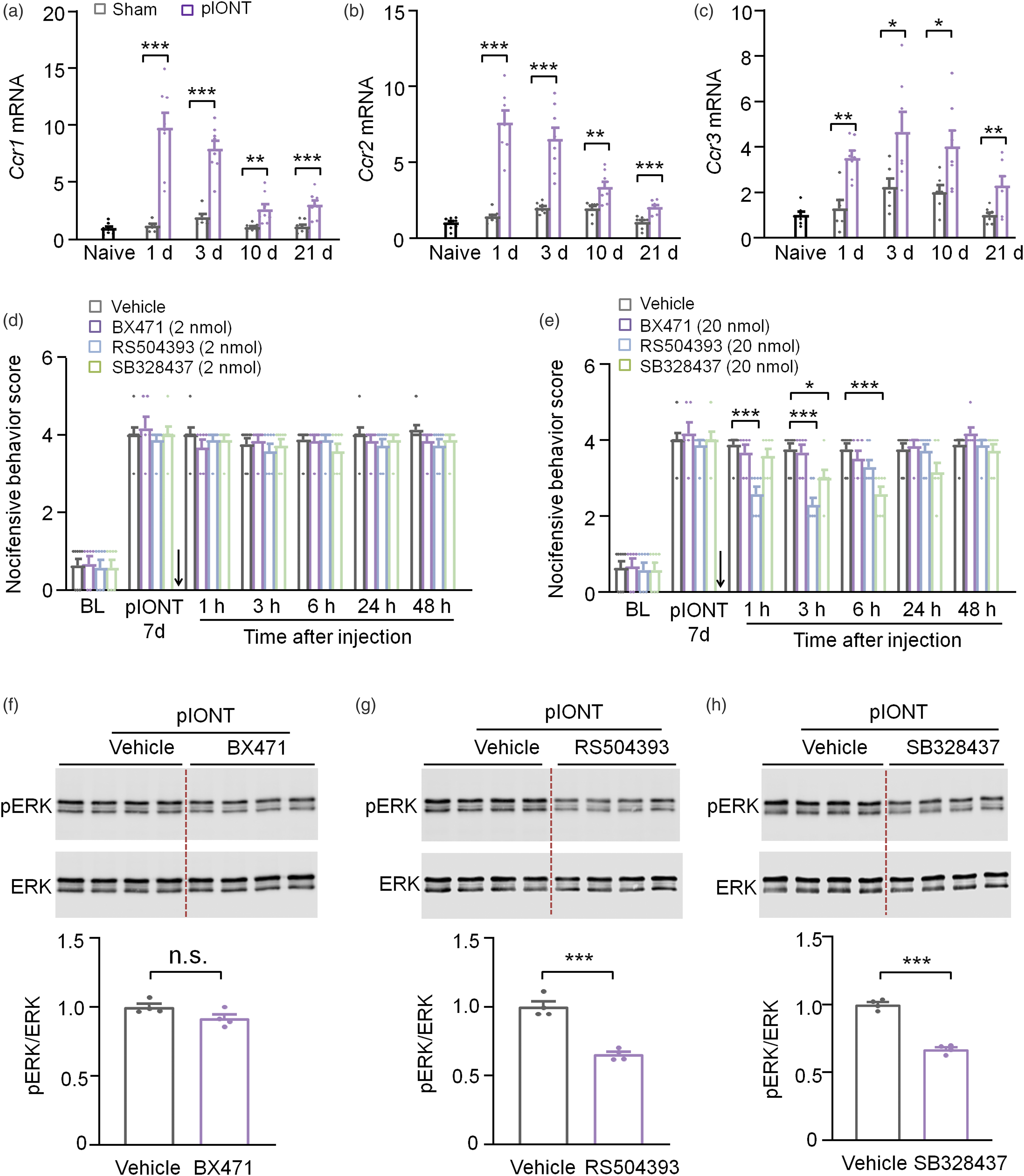
CCR2 and CCR3 are expressed in TG neurons
We further checked the expression and distribution of CCR2 and CCR3 by immunostaining. The CCR2-IR was low in the TG of naive and sham-operated mice, but was increased after pIONT (Figures 5(a)–(d)), confirming the upregulation of CCR2 by pIONT. In addition, double staining showed that CCR2 was colocalized with TUJ1 (Figures 5(e)–(h)), indicating the neuronal expression in the TG. Meanwhile, CCR3 was also increased after pIONT (Figures 5(i)–(l)) and highly colocalized with TuJ1 (Figures 5(m)–(p)). These results suggest that CCR2 and CCR3 are distributed in TG neurons. CCR2 and CCR3 are expressed in TG neurons. (a–c) Representative images of CCR2 immunofluorescence in the TG. CCR2-IR was low in naive mice (a) and sham mice (b), but increased in the TG of pIONT mice (c). (d) The percentage of CCR2+ neurons in naïve, sham, and pIONT groups. ***p < .001, one-way ANOVA followed by the Bonferroni’s test. N, naïve; S, sham. (e–g) Double staining of CCR2 (d) and neuronal marker TUJ1 (e) showed the neuronal expression of CCR2. (h) The percentage of CCR2/TuJ1+ double-labeled neurons in CCR2+ neurons. (i–k) Representative images of CCR3 immunofluorescence in the TG. CCR3-IR was low in naive mice (i) and sham mice (j), but increased in the TG of pIONT mice (k). (l) The percentage of CCR3+ neurons in naïve, sham, and pIONT groups. ***p < .001, one-way ANOVA followed by the Bonferroni’s test. N, naïve; S, sham. (m–o) Double staining of CCR3 (m) and neuronal marker TUJ1 (n) showed the neuronal expression of CCR3. (p) The percentage of CCR3/TuJ1+ double-labeled neurons in CCR3+ neurons.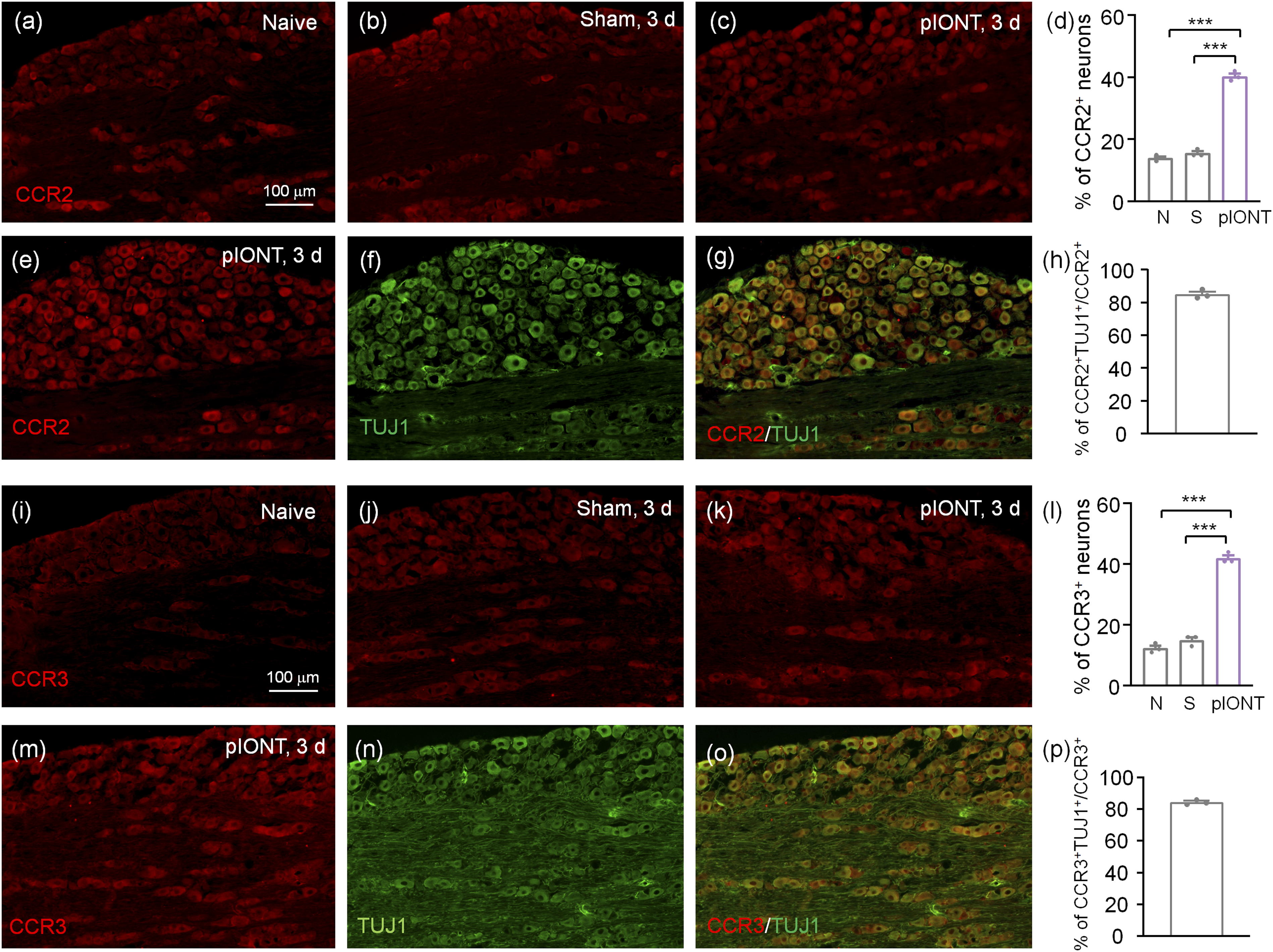
RS504393 and SB328437 reduced CCL7-induced neuronal hypersensitivity
To investigate whether CCL7 enhances TG neuronal excitability by binding CCR2 and CCR3, the TG neurons were preincubated with RS504393 (25 μM) or SB328437 (25 μM) 20 min before CCL7. Whole-cell current-clamp recordings showed that pretreatment with RS540393 significantly increased the rheobase of TG neurons compared with CCL7 only group (Figures 6(a) and (b)). The numbers of APs in response to 160 pA, 1000 ms current injection in the RS504393- and SB328437-pretreated neurons was significantly decreased (Figures 6(c) and (d)). In addition, the APs RMP, S-Rise of TG neurons was significantly decreased and the APs threshold, and half-width was significantly increased in the RS504393-pretreated TG neurons (Figures 6(e)–(h)). The APs threshold of TG neurons in SB328437-pretreated neurons was also significantly increased compared with the CCL7-treated (Figure 6(g)). In addition, parameters such as APs amplitude, medium AHP, s-Decay, and fast AHP were not significantly changed (Figures 6(i)–(l)). These data suggested that CCR2 antagonist and CCR3 antagonist reduced CCL7-induced neuronal hypersensitivity. CCR2 antagonist and CCR3 antagonist reduced CCL7-induced neuronal hypersensitivity. (a) The representative traces of APs evoked by a rheobase current from CCL7-, CCL7+ RS504393-, and CCL7+ SB328437-treated TG neurons. (b) RS504393 increased the rheobase current required to evoke an AP compared with vehicle group. ***p < .001 vs. vehicle; one-way ANOVA followed by the Bonferroni’s tests. n = 16–19 neurons/group from 3-4 mice. (c) The representative traces of APs evoked by depolarizing current steps recorded from CCL7-, CCL7+ RS504393- and CCL7+ SB328437-treated TG neurons. (d) RS504393 and SB328437 decreased the number of APs in response to 160 pA, 1000 ms current injection. ***p < .001, RS504393 vs. vehicle; *p < .05, SB328437 vs. vehicle. Two-way RM ANOVA followed by Bonferroni’s tests. (e) RS504393 increased the resting membrane potential of TG neurons. *p < .05 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (f) RS504393 and SB328437 increased the APs threshold of TG neurons. ***p < .001, RS504393 vs. vehicle. *p < .05, SB328437 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (g) RS504393 decreased the APs s-rise of TG neurons. ***p < .001 vs. vehicle. One-way ANOVA followed by the Bonferroni’s tests. (h) RS504393 increased the APs half-wide of TG neurons. **p < .01 vs. vehicle; one-way ANOVA followed by the Bonferroni’s tests. (i–l) No difference in the APs amplitude (i), medium AHP (j), s-Decay (kj), and fast AHP (l) between CCL7-, CCL7+ RS504393-, and CCL7+ SB328437-treated TG neurons. One-way ANOVA followed by the Bonferroni’s tests.
Discussion
In the present study, we report the function of CCL7 in regulating TG neuronal excitability and trigeminal neuropathic pain. We found that CCL7 was increased in TG neurons after pIONT, and specific inhibition of CCL7 in the TG effectively alleviated pIONT-induced orofacial mechanical allodynia. Furthermore, CCL7 enhanced the excitability of TG neurons and increased the phosphorylation of ERK in the TG. Moreover, pIONT induced the upregulation of CCR1, CCR2, and CCR3 in the TG. However, intra-TG injection of specific antagonists of CCR2 or CCR3, but not of CCR1, attenuated pIONT-induced orofacial mechanical allodynia, reduced ERK activation, and decreased CCL7-induced TG neuronal hyperexcitability. Thus, our results indicate that CCL7 mediates trigeminal neuropathic pain via CCR2/CCR3-ERK pathway in TG neurons.
CCL7 is up-regulated in TG neurons and contributes to the development and maintenance of trigeminal neuropathic pain
Previous studies have shown that trigeminal nerve injury changes the expression of several chemokines in the TG including CCL2, CXCL1, and CXCL13.9–11 Here we show that Ccl7 was persistently up-regulated in the TG. Furthermore, CCL7 is mainly distributed in NF200+ large myelinated type A fiber mechanoreceptors and also distributed in CGRP+ and IB4+ nociceptive neurons. As chemokines are secreted proteins, the increased CCL7 can be released from these neurons and acts on adjacent neurons or non-neuronal cells. Previous studies showed that CCL7 was mainly identified in astrocytes in the spinal dorsal horn after CCI or pSNL,5,42 indicating the different cellular distribution of CCL7 in different tissues.
As important inflammatory mediators, chemokines are key components of chronic disease pathology. 43 CCL7 plays a role in the inflammatory response by attracting macrophages and monocytes to further amplify the inflammatory process and promote the progression of cardiovascular disease, diabetes, and kidney disease. 14 Several studies have shown that CCL7 can promote tumor invasion and metastasis; however, other studies have suggested a tumor suppressive role for CCL7. 44 In pain-related research, Imai et al. 5 demonstrated that a single intrathecal injection of CCL7 induces mechanical allodynia and heat hyperalgesia, whereas an intrathecal injection of blocking antibody to CCL7 inhibits pSNL-induced neuropathic pain . Our behavioral data demonstrated that intra-TG injection of siRNA targeted CCL7, soon after pIONT as well as 7 days after the procedure effectively alleviated the pIONT-induced mechanical allodynia. Intra-TG injection of CCL7 recombinant protein induced orofacial mechanical allodynia in naïve mice with a dose-dependent manner. Similarly, application of CX3CL1, CCL2, CXCL1, CXCL13, or CXCL10 to TG resulted in orofacial mechanical allodynia,9–11,34,45,46 suggesting that chemokines are important mediators in the pathogenesis of trigeminal neuropathic pain.
CCL7 mediates trigeminal neuropathic pain via CCR2/CCR3-ERK cascade in TG neurons
Chemokines exert their biological effects through G protein-coupled receptors.35,47 CCR1, CCR2, and CCR3 are known as the functional receptors of CCL7.14,26 Pawlik et al. 48 reported that CCL2/7/8/CCR1 and CCL7/8/CCR3 signaling are important in the modulation of neuropathic pain. We show that intra-TG injection of specific antagonist of CCR2 or CCR3, but not of CCR1, reversed the mechanical allodynia induced by pIONT. Consistently, specific antagonist of CCR2 or CCR3 alleviated CCL7-induced hyperexcitability of TG neurons. Furthermore, CCR2 and CCR3 were persistently up-regulated in the TG neurons after pIONT, indicating that CCL7 and these receptors in the TG mediate trigeminal neuropathic pain via neuron-neuron interaction. CCL2-CCR2, CXCL10-CXCR3, and CXCL13-CXCR5 are also involved in trigeminal neuropathic pain via the similar way.9–11 A previous study demonstrated that CCL7 is produced by spinal astrocytes and acts on CCR2 in microglia, thereby inducing spinal microglial activation and contributing to sciatic nerve injury-induced neuropathic pain. 5 Differently, nerve injury increases CXCL10 and CXCR3 in spinal astrocytes and neurons, respectively, 49 and CXCL13 and CXCR5 in spinal neurons and astrocytes, respectively. 7 Thus, chemokine and receptor pairs mediate neuropathic pain via different mechanisms in different tissues.
It is well known that MAPKs, including ERK, p38, and JNK are activated in the DRG and spinal cord by peripheral nerve injury. 32 In trigeminal neuropathic pain models, ERK, p38, and JNK are activated and contribute to mechanical allodynia.10,11,24,33,34 Inhibitor of p38 or ERK attenuated pIONL-induced pain by reducing the expression of pro-inflammatory mediators such as TNF-α and IL-1β in the TG.10,34 Here we show the activation of ERK by CCL7 via CCR2 and CCR3. A range of chemokines, including CCL2, CXCL10, and CXCL13, binding with receptor leads to the activation of ERK,10,11,50 whereas intrathecal CXCL9 or CXCL11 did not induce ERK phosphorylation. 51 Certainly, ERK is not always expressed in neurons. CXCL13 induces ERK-dependent astrocytic activation in the spinal cord and pain hypersensitivity. 7 In addition to ERK, phosphorylated AKT is up-regulated after CCR2 or CCR3 activation.52,53 Up-regulation and activation of CCR1, CCR2, and CCR5 activate the associated JAK/STAT1/STAT3 pathway at different stages of a model for rheumatoid arthritis. 54 Thus, CCL7 may also act through other downstream pathways to contribute to neuropathic pain.
CCL7-CCR2/CCR3 is involved in regulating neuronal hyperexcitability after pIONT
Aberrant hyperexcitability of sensory neurons is a key contributor of chronic pain. 41 Voltage gated sodium channels (VGSCs) play a key role in regulating neuronal excitability. 55 Chemokines including CCL2, CXCL1, and CXCL13 modulate Na+ currents in DRG neurons by increasing the expression of Nav1.7 and Nav1.8 or increasing current density.8,56–58 CCL2 promotes peripheral sensitization by acting on the biophysical properties of Nav1.8 currents via a CCR2/Gβγ-dependent mechanism. 59 Similarly, CXCL12 increased Nav1.8 expression in an ERK-dependent manner and triggered intracellular calcium influx in Nav1.8-positive DRG neurons, leading to hyperexcitability of DRG neurons.60,61 Besides VGSCs, ligands of CXCR2 participated in mechanical allodynia via down-regulation of voltage-gated potassium channels (VGPCs, such as Kv1.4 and Kv1.1) 45 or TRPV1 channels. 62 In addition, ERK plays a role in the modulation of VGPCs, such as Kv4.2 and Kv4.3.63,64 We showed that CCL7 increased hyperexcitability in small-diameter nociceptive TG neurons, which was reduced by CCR2/CCR3 antagonist. As CCL7 also activates ERK, it is possible that CCL7-CCR2/CCR3 excites primary sensory neurons and peripheral sensitization by regulating the function of sodium channels or potassium channels, which needs further investigation in the future.
In summary, we provided the first evidence that CCL7, CCR2 and CCR3 in the TG neurons were involved in pIONT-induced orofacial mechanical allodynia. Our results demonstrated that CCR2 and CCR3 can be activated by CCL7 and induces downstream ERK activation, which increases neuronal excitability and further contributes to the maintenance of neuropathic pain. Thus, targeting the CCL7-CCR2/CCR3-ERK pathway in the TG may provide a novel therapeutic approach for the treatment of the trigeminal neuropathic pain.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the STI2030-Major Projects (2022ZD0204700); National Natural Science Foundation of China (32030048); National Natural Science Foundation of China (32200817), National Natural Science Foundation of China (82271256); and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX21_3135).