
Abstract
GABAergic system disinhibition played an important role in the pathogenesis of remifentanil-induced hyperalgesia (RIH). K+-Cl--cotransporter-2 (KCC2) has the potential to enhance the strength of GABAergic signaling function. However, few reports have focused on the additive analgesic effect of KCC2 enhancer and GABAA receptor agonist on the spinal dorsal horn. Therefore, we evaluated the role of GABA type A receptor (GABAAR) agonist (muscimol), KCC2 enhancer (CLP257) in remifentanil-induced hyperalgesia, as well as GABA and KCC2 receptors responses in the dorsal spinal horn. Remifentanil started to reduce paw withdrawal mechanical thresholds at postoperative 4 h and lasted to 72 h. The RIH associated decreases in spinal GABA release was transient. The amount of spinal GABA transmitter by microdialysis was observed to be decreased at the beginning and reached bottom at 150 min, then returned to the baseline level at 330 min. The synthesis and transportation of GABA transmitter were inhibited, characterized as spinal GAD67 and GAT1 downregulation after the establishment of RIH model. The effect of RIH on GABA receptor downregulation was linked to the reduced expression of spinal KCC2 receptor. This decrease in KCC2 expression has coincided with an early loss of GABA inhibition. KCC2 enhancer, which is reported to lead to a reduction in intracellular Cl−, can enhance GABA-mediated inhibitory function. Both muscimol and CLP257 could dose-dependently inhibit mechanical hypersensitivity caused by remifentanil-induced downregulation of GABAAα2R and KCC2, respectively. Compared with muscimol acting alone, the joint action of CLP257 and muscimol showed a higher pain threshold and less c-fos expression via upregulation of KCC2 and GABAAα2R. Taken together, these findings suggested that the RIH was initiated by decreased GABA release. Downregulation of GABAAα2R and KCC2 receptor contributed to spinally mediated hyperalgesia in RIH. KCC2 enhancer was proved to potentiate antinociceptive effect of GABAAR agonist in RIH.
Introduction
Remifentanil is commonly used for alleviating intraoperative surgical pain. However, remifentanil-induced hyperalgesia (RIH) has become a major public concern in recent years. 1 Opioid, especially remifentanil, can paradoxically stimulate the pronociceptive systems and cause unexpected allodynia. 2 Many reports demonstrated that remifentanil exposure exacerbated postoperative nociceptive sensitization, rather than antinociceptive effect.3,4 Although remifentanil is an ultra-short-acting agent, it could induce delayed and sustained hyperalgesia lasting for 2–3 days.
γ-aminobutyric acid (GABA), one of the major inhibitory neurotransmitters, is released by neurons in the spinal dorsal horn (SDH). Under normal conditions, spinal activation of GABAergic neurons was essential in the spinal antinociceptive circuits. 5 Disinhibition of GABAergic systems has been proposed as the important mechanism in central sensitization initiation and hyperalgesia.6,7 It was confirmed that RIH was initiated by inhibiting GABA receptors on the neurons in SDH. 8
GABAergic disinhibition was reported to result from deactivation of spinal K+-Cl−-cotransporter-2 (KCC2) signal.9,10 The KCC2 sets a low intracellular chloride concentration and leads to GABA-mediated postsynaptic inhibition. 11
Opioid was reported to inhibit KCC2 function and impair intracellular chloride homeostasis in spinal lamina I neurons, which attributed to hyperalgesia. 7 However, there are no reports yet that involved the GABA release and GABA, KCC2 receptor regulation in the research of RIH prevention.
We therefore extended our hypothesis to address whether RIH could decrease spinal KCC2 expression followed by an unanticipated downregulation of spinal GABA function. Moreover, whether the KCC2 enhancer could have synergistic augmentation in RIH rats treated with GABAAR agonist. This study was conducted to investigate the additive effect of KCC2 enhancer with GABAAR agonist in RIH model rats.
Methods
Animals
Male Sprague-Dawley rats were provided by the department of experimental animal sciences, Wenzhou Medical University. Rats were 8–10 weeks old and weighed 220–250 g at the time of surgery. All rats had an acclimation period of at least 7 days with food and water freely available. The rats were maintained at a constant temperature (23 ± 0.5°C), humidity (55–60%) and irradiation (12:12 h light: dark cycle). The studies were performed in accordance with Guide for the Care and Use of Laboratory Animals. This manuscript adheres to the ARRIVE guidelines. The procedures involved in the study were approved by the animal ethical care committee of Wenzhou Medical University (No. WYDW2019-0549).
Intrathecal catheter insertion
Under sevoflurane anaesthesia, a sterilized polyethylene-10 (PE - 10) tube (ID 0.28 mm, OD 0.61 mm, Smiths Medical, UK) was implanted between the L3–4 intervertebral space. Experiments were performed 7 days after intrathecal catheter implantation. Rats showing any neurological dysfunction were excluded.
Rat model of remifentanil-induced hyperalgesia (RIH model) and surgical incision
Under sevoflurane anaesthesia, a 24-gauge catheter (BD Angiocath, Utah, USA) flushed with heparinized saline was placed into the caudal vein. Remifentanil was infused at a rate of 1.2 μg · kg−1 · min−1 for 60 min. Meanwhile, a longitudinal incision, starting 0.8 cm from the proximal edge of the heel extending towards the toes, was made in the left hindpaw to mimic surgical stress. After hemostasis, the incision was ligated and covered with antiseptic gauze. All of the procedures were performed under sevoflurane anaesthesia.
Test drugs
Remifentanil (90A03111, Yichang Humanwell Pharmaceutical Co. Ltd., Hubei, China) was prepared with normal saline for intravenously infusion through the tail vein by 24 G catheter. Both muscimol (M1523, Sigma-Aldrich, St Louis, MO, USA, dissolved in 0.05 M HCl) and CLP257 (HY-110,143, MedChemExpress, Monmouth Junction, NJ, USA, dissolved in 1% DMSO) were intrathecally injected through intrathecal catheter. The rats were administrated under sevoflurane anaesthesia 30 min prior to remifentanil infusion.
Pain assay
Paw withdrawal mechanical thresholds (PWMT) were evaluated using von Frey filaments (Aesthesio Co., USA). Rats were placed in separated transparent plastic cages (10 × 20 × 9 cm) with a wire mesh bottom (1 × 1 cm). The von Frey filaments were used to apply a bending force (range from 1 g to 26 g to avoid unexpected injury) to the plantar surface of the hindpaw. The PWMT was determined by the “up-and-down” method. Paw licking or withdrawal was considered nociceptive-like response.
The pain threshold was calculated according to the formula: P
All PWMT results were presented as a percentage of the basal PWMT value. Each rat was allowed to acclimatize for 30 min before testing. The research fellows in behavioural experimenters were blinded to the groupings of the rats.
Muscimol and CLP257 are sedative, and therefore will inherently alter PWMT. To exclude the interference effect, assessment of sedation degree should be performed before pain assay. According to the Shimoyama sedative scale, the rats with score exceeded 1 were excluded from the pain assay (0 Normal; 1 Cannot balance on hind limbs; 2 Cannot negotiate 60° vertical mesh; 3 Loss of righting reflex; 4 Immobility (reaction to pain); 5 No reaction to pain). The Shimoyama sedative score of the rats before pain assay was attached in Supplemental Table 1.
Western blot
After the last behavioural test, the rats were euthanized. L4–L6 spinal cords were rapidly harvested and stored in liquid nitrogen. The samples were homogenized in RIPA lysis buffer containing a cocktail of protease inhibitor, phosphatase inhibitors and phenylmethylsulfonyl fluoride (Sigma-Aldrich) protease inhibitors. Then the lysate was centrifuged at 11,000 r/min for 25 min at 4°C and the supernatant was removed. The protein concentration in the supernatant was estimated using a bicinchoninic acid (BCA) protein assay kit (Pierce). Samples (30 μg) were separated on 8% SDS-PAGE gel electrophoresis and transferred onto a PVDF membrane. The filter membranes were blocked with 5% skim milk in TBST for 1.5 h at room temperature and subsequently incubated overnight at 4°C with the following primary antibodies: glutamic acid decarboxylase 65 (GAD65) (1: 5000, ab239372, Abcam),γ-aminobutyric acid transporter-1 (GAT1) (1: 500, ab426, Abcam), KCC2 (1: 1000, ab49917, Abcam), phospho-Ser940KCC2 (1: 1000, PhosphoSolutions, Aurora, CO, USA), c-fos (1: 2000, ab190289, Abcam), GAPDH (1: 1000, 5174, Cell Signaling Technology), glutamic acid decarboxylase 67 (GAD67) (1: 1000, ab26116, Abcam) and γ-aminobutyric acid type A receptor α2 subunit (GABAAα2R) (1: 500, ab193311, Abcam)). The membrane was washed with TBST and followed by incubation with the goat anti-mouse (1: 10,000, BL001A, Biosharp) or goat anti-rabbit secondary antibody (1: 10,000, BI003A, Biosharp) conjugated with horseradish peroxidase for 1 h at room temperature. The density of the blots was detected by using a chemi-Dos XRS imaging system and quantified by Image J software (Syngene, Cambridge, UK) and normalized against endogenous control (GAPDH) immunoreactivity.
In vivo microdialysis
Under sevoflurane anaesthesia, a guide cannula was unilaterally implanted into the L1 spinal dorsal horn, with a 30-degree angle cephalad from the horizontal plane. At the end, guide cannula was stabilized with dental cement. After a recovery period of 24 h, rats showing any neurological dysfunction were excluded. A microdialysis probe (CMA 12 elite metal free, CMA microdialysis, Sweden) was inserted into the guide cannula, leaving the active dialysis membrane (length 1 mm, Polyarylethersulfone, OD 0.5 mm, molecular cutoff 20,000 Da) inside.
The dialysis system was attached to a peristaltic pump (CMA 402 syringe pump) and perfused with artificial cerebral spinal fluid (124 mM NaCl, 2.5 mM KCl, 1.0 mM MgCl2, 2.0 mM CaCl2, 1.25 mM NaH2PO4, 10.0 mM glucose and 26 mM NaHCO3, Coolaber, Beijing, China) at a flow rate of 1 μL/min for a 90-min stabilization period before remifentanil infusion. Then three consecutive 30-min samples were collected as baseline values.
To eliminate the surgical wound’s interference, the rats involved in this part were only infused with remifentanil without surgical incision. At the beginning of the remifentanil administration, samples were collected at 30-min intervals for 480 min and frozen at −80°C until analysed for GABA, within 24 h. At last, the rats were sacrificed and the sections (30 μm) were stained with cresyl violet to identify the location of the catheters. Only the rats with the microdialysis probe placed in SDH region were included for analysis.
Determination of GABA
The concentration of GABA was detected by LC-MS/MS Analysis (Triple Quad 4500 MD, AB Sciex, USA). Chromatography was performed on a Kinetex® 2.6 μm C18 100 Å column with the diameter of 2.1 × 100 mm (00D-4462-AN, phenomenex, USA), using 30:70 (v/v) acetonitrile-water containing 5 mM ammonium acetate. The temperature of column was 40°C under a flow rate of 0.3 mL/min. The sample injection was 5 μL, and run-to-run time was 2 min. Positive ion multiple-reaction monitoring (MRM) mode with 5500 eV ion spray voltage, 500°C heated capillary temperature, 50 psi sheath gas (nitrogen) and 50 psi auxiliary gas (nitrogen) was used to detect the GABA (m/z 104/87.1). All results were presented as a percentage of the basal amount of GABA, which was calculated as the average of the three baseline values.
Experimental protocol
Experiment 1: Behavioural test, WB analysis and in vivo SDH microdialysis in RIH rats
Forty two rats were randomly divided into seven groups (n = 6): Group C (Control Group with surgical incision); Group 4 h (4 h after RIH model); Group 8 h (8 h after RIH model), Group 16 h (16 h after RIH model), Group 24 h (24 h after RIH model), Group 48 h (48 h after RIH model) and Group 72 h (72 h after RIH model). PWMT tests were measured at −24 h (baseline value), 4 h, 8 h, 16 h, 24 h, 48 h and 72 h after remifentanil infusion. The proportion of PWMT change after remifentanil infusion comparing with −24 h was recorded. After the behavioural tests, the expressions of GAT65, GAT67, GAT1, GABAAα2R and KCC2 were measured by using Western Blot analysis. A schematic diagram of this part was shown in Figure 1(a). A–C schematic diagram in experiments 1–3. (A) Behavioural change and protein expression after remifentanil exposure in RIH rats. (B) In vivo spinal dorsal horn microdialysis after remifentanil exposure in rats. (C) Additive potentiation by co-application of KCC2 enhancer and GABAAR agonist in inhibiting RIH.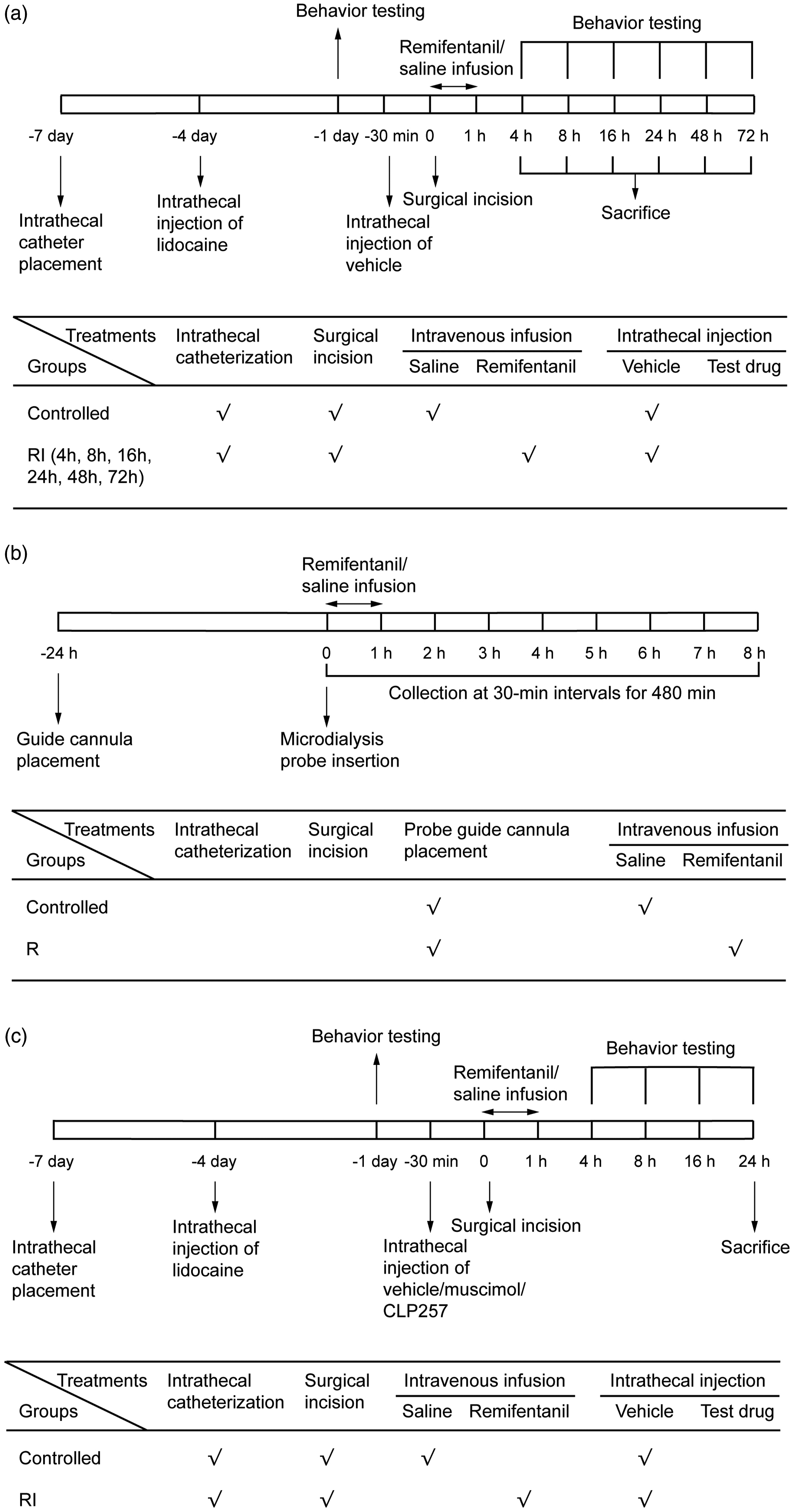
Twelve rats received remifentanil infusion or normal saline (n = 6) without surgical incision. Samples used for determination of GABA were collected at 30-min intervals from the beginning of remifentanil infusion to 480 min. A schematic diagram of this part was shown in Figure 1(b).
Experiment 2: Dose-dependent effect of KCC2 enhancer (CLP257) or GABAAR agonist (muscimol) in inhibiting RIH
Forty eight rats were randomly divided into the eight groups (n = 6): Group C received normal saline infusion with surgical incision; Group RI received remifentanil infusion and surgical incision. The following groups were performed based on the group RI. Group Mus-low received 0.1 μg muscimol; Group Mus-medium received 0.3 μg muscimol; Group Mus-high received 0.9 μg muscimol; Group CLP-low received 1 μg CLP257; Group CLP-medium received 10 μg CLP257; Group CLP-high received 100 μg CLP257. PWMT tests were measured at −24 h (baseline value), 4 h, 8 h, 16 h and 24 h after remifentanil infusion. After the last behavioural test, the expressions of GABAAα2R and KCC2 were measured by using Western Blot analysis.
Experiment 3: Additive potentiation by co-application of KCC2 enhancer and GABAAR agonist in inhibiting RIH
Thirty rats were randomly divided into the five groups (n = 6): Group C received normal saline infusion with surgical incision; Group RI received remifentanil infusion and surgical incision. Rats in the following four groups were performed based on the operation in Group RI. Besides remifentanil infusion and surgical incision, Group Mus received 0.3 μg muscimol, Group CLP received another 10 μg CLP257; Group MC received 10 μg CLP257 and 0.3 μg muscimol.
PWMT tests were measured at −24 h (baseline value), 4 h, 8 h, 16 h and 24 h after remifentanil exposure. After the last behavioural test, the expressions of c-fos, GABAAα2R, KCC2 and phospho-Ser940KCC2 were measured by using Western Blot analysis. A schematic diagram of this part was shown in Figure 1(c).
Statistical analysis
Sample size calculation and statistical analysis were conducted using PASS 15.0 and SPSS 20.0, respectively. The proportion of the PWMT change at 24 h comparing with −24 h in Group RI (primary outcome) was 85.8%, whereas the data of the Group C and three treatment groups (Group CLP, Group Mus and Group CM) were 0.5%, 66.1%, 54.3% and 17.8%, respectively. Assuming an α error of 0.05 with a power of 0.80, a sample size of six rats per group was required to identify a significant effect. Data were expressed as means ± standard deviation (SD). Hyperalgesia-related behavioural tests and GABA release among the groups at different time points were compared using the repeated measures analysis of variance (ANOVA). The results showed that there were interactions between time points and PWMT change and GABA release, respectively, so we used one-way ANOVA to assess the differences in PWMT change at each time point. GABA release was compared using independent t-test. In addition, the expression of GAT65, GAT67, GAT1, GABAAα2R, KCC2 and c-fos among the groups was compared using one-way ANOVA followed by LSD post hoc analysis. A p-value less than 0.05 was considered statistically significant.
Result
Remifentanil exposure induced downregulation of GABAAα2R and KCC2 as well as inhibition of GABA release
Rats in RIH groups displayed significant mechanical hyperalgesia compared with the controlled group. As shown in Figure 2, the right hindpaw without surgical incision in Group RI showed significant remifentanil-induced hyperalgesia, which started at 4 h (Right hindpaw in Control vs RI: 1.71 ± 7.1% vs −45.1 ± 15.3%, p < 0.001) to 8 h (Control vs RI: −0.56 ± 3.5% vs −57.73 ± 13.1%, p < 0.001), and reached to peak at 24 h (Control vs. RI: −1.19 ± 3.7% vs. −84.23 ± 6.8%, p < 0.001) and 48 h (Control vs RI: 5.01 ± 10.3% vs −85.92 ± 5.9%, p < 0.001) after the first mechanical threshold test at −24 h (the same below). Interestingly, there was an additional peak of pain outbreak (4 h–8 h) after establishment of RIH in the left hindpaw with surgical incision in Group RI. Compared with remifentanil infusion alone in the right planter, surgical incision in the left planter can exacerbate the existing RIH from 4 h (Left vs Right: −90.40 ± 4.4% vs −45.1 ± 15.3%, p < 0.001) to 8 h (Left vs Right: −90.05 ± 4.2% vs −57.73 ± 13.1%, p < 0.001) in Group RI (Figure 2). PWMT change after remifentanil infusion. Remifentanil with (right hindpaw)/without (left hindpaw) surgical incision decreased the value of PWMT at different time courses. Data were expressed as percent change from the baseline (mean ± SD) (n = 6). **p < 0.01 versus group C; &&p < 0.01 versus group RI (Right hindpaw).
As shown in Figure 3, the spinal GABA level was gradually depressed after remifentanil exposure. The amount of GABA was observed to be decreased at the beginning of remifentanil exposure (at 30 min) (p = 0.002, 31.50 [15.18–47.81]) and reached the bottom at 150 min (p < 0.001, 82.65 [66.49–98.81]), then returned to baseline at 330 min (p = 0.052, 19.11 [−0.19–38.41]). Changes of GABA concentration in SDH after remifentanil exposure. Open squares and closed circles indicate GABA concentrations in the rats with administration of intravenous remifentanil or normal saline, respectively (n = 6). Grey bar: the interval of remifentanil infusion. Data were presented as percent change from the basal concentration (mean ± SD) (n = 6). *p < 0.05 or **p < 0.01 versus group C.
Compared with group C, the expression of GABAAα2R in group RI was suppressed at 16 h (p = 0.015, 95% CI [0.03–0.25]) and 24 h (p = 0.019, 95% CI [0.02–0.24]). Of note, GAD67 and GAT1, which play roles in the synthesis and transportation of GABA neurotransmitter, were inhibited at the beginning of the remifentanil exposure, lowest at 4 h (p < 0.001, 95% CI [0.23–0.50]) and 8 h (p = 0.038, 95% CI [0.02–0.51]), respectively. The expression of KCC2 in group RI began to be suppressed at 24 h (p = 0.011, 95% CI [0.06–0.36]), and down to lowest at 48 h (p = 0.008, 95% CI [0.07–0.38]) (Figure 4). Effects of remifentanil exposure at different time courses in lumber spinal cord. Western blotting showed that the expression of GAT1 (8 h), GAD67 (4 h, 8 h, 24 h), KCC2 (24 h, 48 h) and GABAAa2R (16 h, 24 h) was down-regulated in spinal cord after remifentanil infusion, and GAD65 remained unchanged. GAPDH was used as a loading control. (A) Expression of GAD65, GAD67, GAT1, GABAAα2R and KCC2 at different time courses. (B) The fold change for the density of GAD65, GAD67, GAT1, GABAAα2R and KCC2. Data were expressed as mean ± SD. *p < 0.05 or **p < 0.01 versus group C.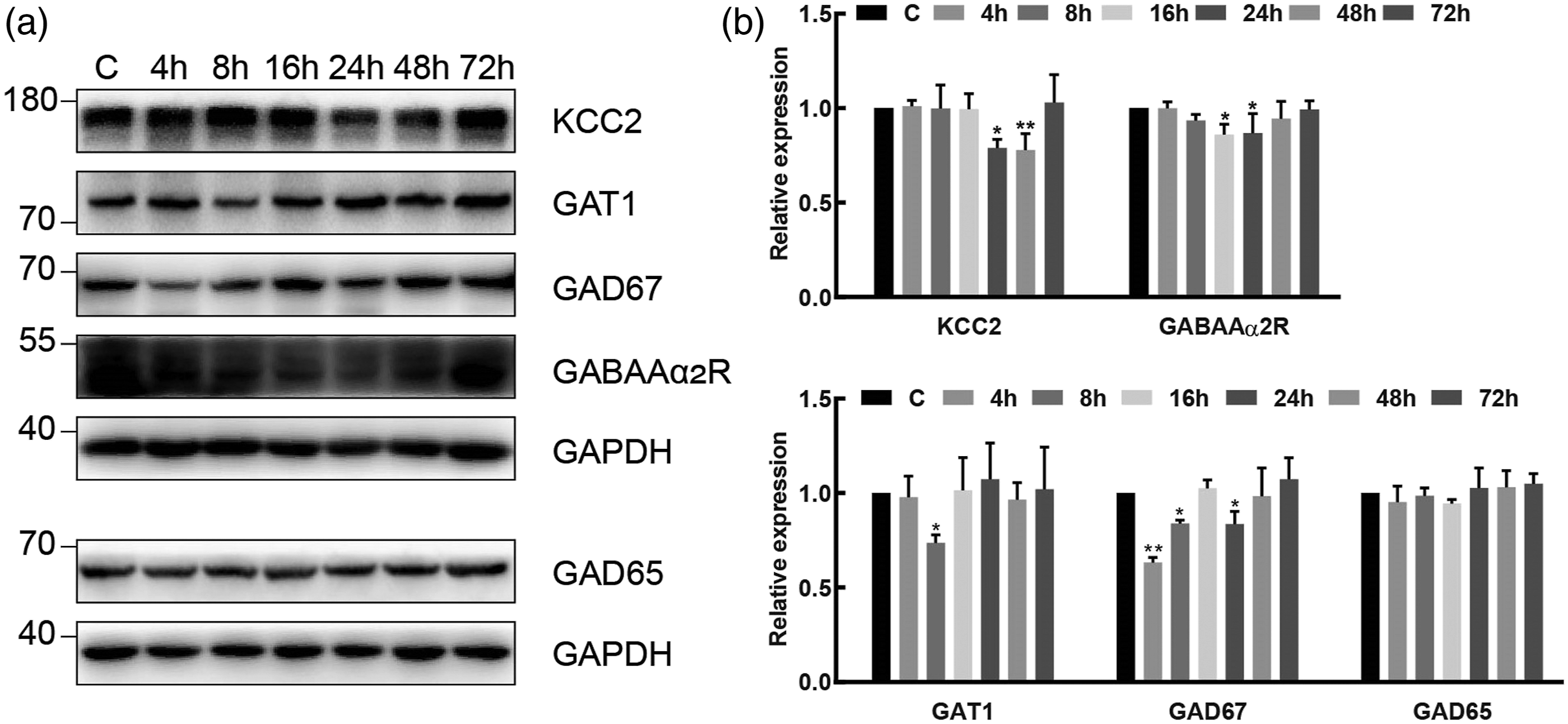
CLP257 and muscimol dose-dependently inhibit hyperalgesia in RIH rats
To determine the involvement of GABA and KCC2 in RIH, different doses of CLP257 or muscimol were intrathecally administrated 30 min prior to remifentanil infusion. As shown in Figure 5, pretreatment with muscimol or CLP257 dose-dependently effectively attenuated mechanical allodynia, accompanied by upregulating GABAAα2R and KCC2 expression, respectively. It was suggested that activation of GABAAα2R or KCC2 receptor participates in RIH. Muscimol or CLP257 dose-dependently inhibits RIH. (A, B) PWMT were evaluated at −24 h (baseline) and 4 h, 8 h, 16 h and 24 h after remifentanil administration. Data were expressed as percent change from the baseline (mean ± SD) (n = 6). (C, E) Bands of western blot for the expression of GABAAα2R or KCC2 at 24 h after remifentanil administration. (D, F) The fold change for the density of GABAAα2R or KCC2 normalized to the GAPDH level. Data were expressed as mean ± SD (n = 4). **p < 0.01 group C; #p < 0.05 or ##p < 0.01 versus group RI; $p < 0.05 or $$p < 0.01 versus group low; &p < 0.05 or &&p < 0.01 versus group medium.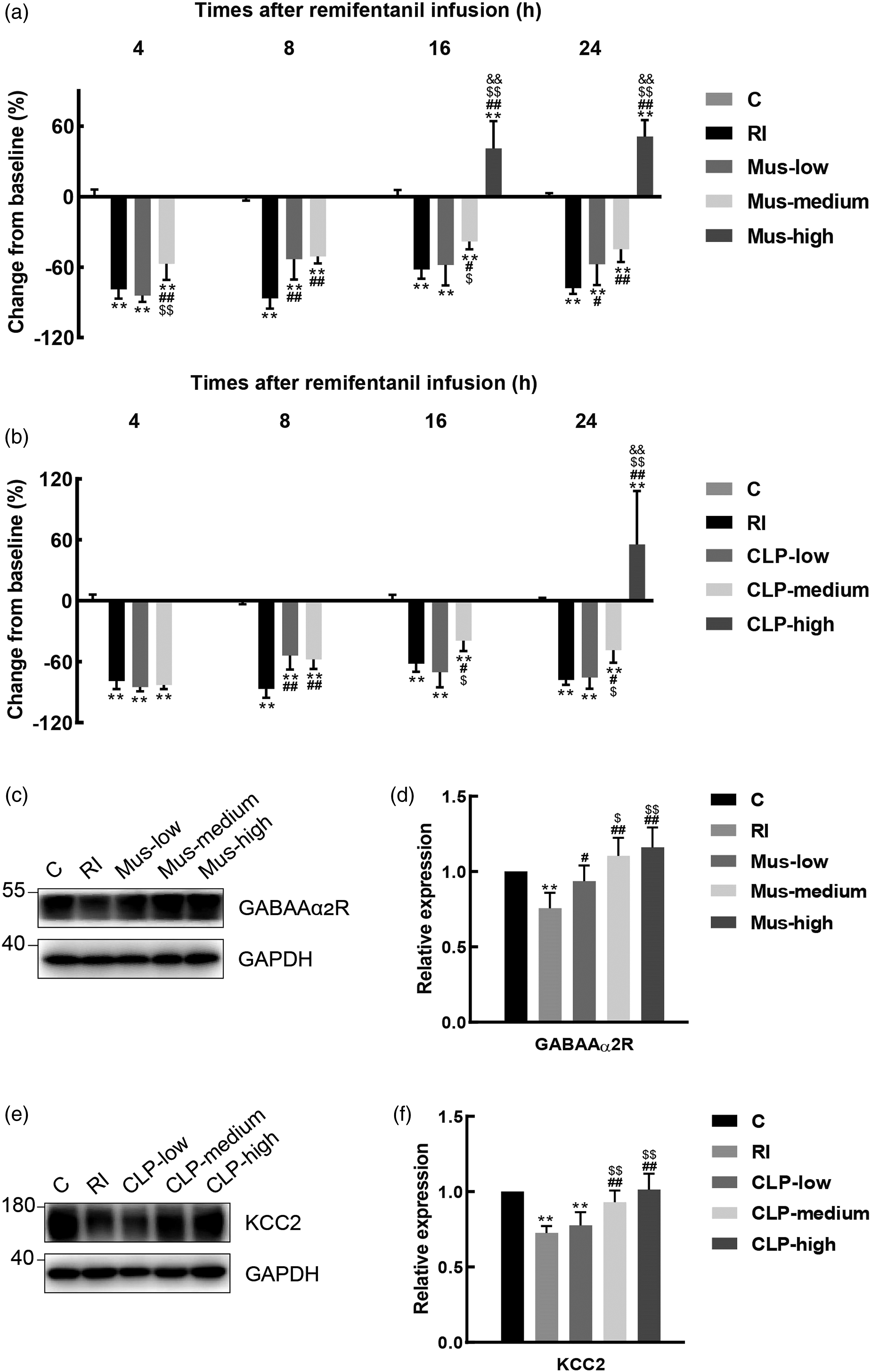
Although the group with high dose of CLP257 or muscimol increased the value of the mechanical threshold, even higher than the control group, most of rats with high doses had transparent paralysis and delayed recovery, which can last for more than 16 h.
The additive effect of CLP257 with muscimol in the reversion of RIH
As shown in Figure 6, compared with treatment of CLP257 (10 μg) or muscimol (0.3 μg) alone, the additive CLP257 with muscimol in Group MC presented higher threshold of PWMT at different points (p < 0.001). Except 4 h after remifentanil infusion (p < 0.001, 95% CI [1.6–3.6]), Group Mus had no potential benefit in attenuating mechanical hyperalgesia in comparison with Group CLP. The additive effects of CLP257 with muscimol in the reversion of pathogenesis of RIH. (A) PWMT was evaluated at −24 h (baseline) and 4 h, 8 h, 16 h and 24 h after remifentanil administration. Data were expressed as percent change from the baseline (mean ± SD) (n = 6). (B) Bands of western blot for the expression of KCC2, pKCC2, GABAAα2R and c-fos at 24 h. (C) The fold change for the density of KCC2, GABAAα2R or c-fos normalized to the GAPDH level. Data were expressed as mean ± SD (n = 4). *p < 0.05 or **p < 0.01 versus group C; #p < 0.05 or ##p < 0.01 versus group RI; $p < 0.05 or $$p < 0.01 versus group Mus; &p < 0.05 or &&p < 0.01 versus group CLP.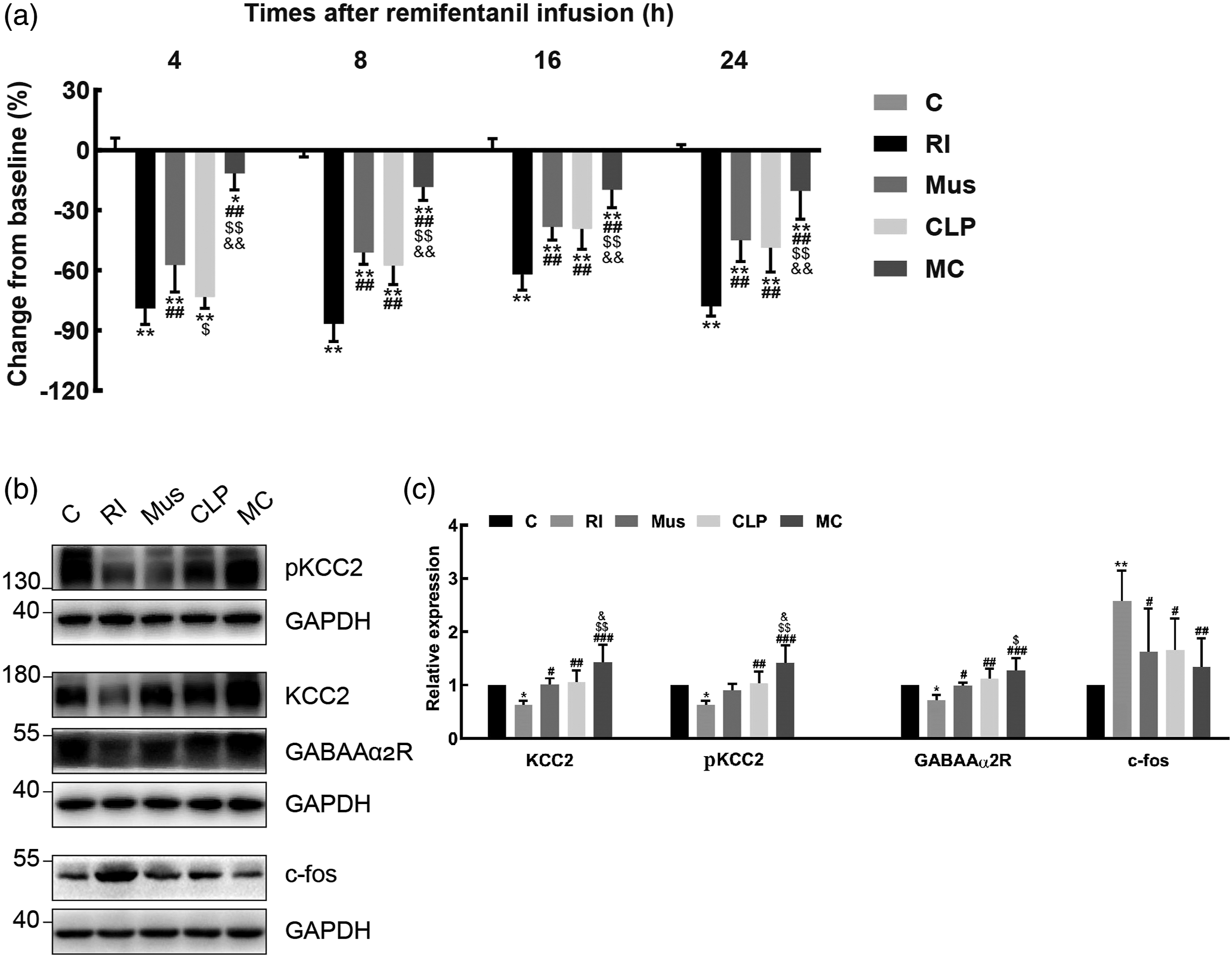
At 24 h after remifentanil exposure, Group MC had more expression in KCC2 (vs CLP, p = 0.013; vs Mus, p = 0.007]), less inhibition of GABAAα2R (vs CLP, p = 0.029; vs Mus, p = 0.010]) and less expression of c-fos (vs CLP, p = 0.032; vs Mus, p = 0.030]).
Discussion
The present study found that: (1) RIH could induce time-dependent downregulation of KCC2, GABAAα2 receptor and its related components in rat lumbar spinal cord. (2) Microdialysis showed that the remifentanil infusion could depress spinal GABA level. (3) Both GABAAR agonist and KCC2 enhancer could dose-dependently inhibit remifentanil-induced behavioural nociception and ameliorate RIH. (4) KCC2 agonist could potentiate the antinociceptive effect by GABAAR agonist.
Of note, surgical incision that induced acute inflammation in the left hindpaw could exacerbate the pre-existing RIH in the early stages (from 4 h to 8 h after incision). At 16 h after RIH establishment, the acute pain threshold caused by surgery returned to the non-surgical level. Despite the surgical incision, the left hindpaw without remifentanil infusion in controlled group showed that the value of PWMT had no statistical difference with the non-surgical site at any time course. The simple surgical incision in the rats without inflammation or neuropathic syndrome was not an independent risk factor to induce postoperative hyperalgesia.
Imbalance of neuronal excitation and inhibition was attributed to neuropathic pain and stress.12,13 NMDA receptor activation in the SDH was proved to be involved in the maintenance of RIH.14,15 GABA, as the main inhibitory neurotransmitter, can counteract NMDA-induced hyperexcitability in the physiological state. 16 Reduced synaptic GABAergic transmission in the spinal cord contributed to an increase in the levels of tactile allodynia and thermal hyperalgesia in rats. 17 Impaired GABAergic inhibitory function may translate behaviourally into the state of hyperalgesia.18,19
Chronic morphine treatment was reported to inhibit spinal GABA release, which could be reversed by acute withdrawal and provided temporarily stimulated increased GABA release. 20 It offered direct evidence that GABA may contribute to the nociceptive response mediated by opioids. Our findings indicated that the GABA release began to decrease after the remifentanil infusion, reached to the bottom level at 150 min and returned to the normal level at 330 min, as followed by the downregulation of the GABAAα2R at 16–24 h after remifentanil infusion in the spinal dorsal horn.
The GABA is mainly synthesized from glutamic acid by GAD67 and GAD65. Decreased amount of GABA release was caused by the inhibition of GAD67. 12 GAT1 has the function to transport GABA from the synaptic cleft of GABAergic axon terminals. 21 In this trial, the remifentanil exposure was proved to impair the neuronal GABA transportation via the blockade of spinal GAT1 (peak at 8th h) expression. The temporary depressed release of GABA was ahead of the impaired GAT1 in the trial. So the early stage of the spontaneously depressed GABA release was not resulted from the GABA transportation by GAT1 downregulation.
GABAAα2R downregulation was delayed inhibited 24 h after remifentanil infusion. GABAAα2R modulation was demonstrated to be involved in the spinal nociceptive circuit.22,23 Enhancement of GABAAα2R by muscimol in the trial has elicited dose-dependently antinociceptive process. It is noteworthy that 0.9 μg of muscimol brought about much more incidence of paralysis and somnolence in the rats. Therefore, the experimental dose of muscimol was set at 0.3 μg.
Previous report about the spinal NMDA receptor overexpression in RIH rats 15 together with the present finding of the downregulation in spinal GABAAα2R clearly indicates that both excitatory and inhibitory neurotransmitter and their receptors modulation are involved in the occurrence and development of RIH. Compared with the pro-nociceptive effect caused by GABA downregulation (peak at 24 h after RIH in this research), the RIH-induced NMDA receptor upregulation was delayed (peak at 48 h14,15). Interestingly, the previous report has proved that the hyperalgesia induced by stress was initiated by a decreased GABA release and maintained by an increased glutamate release at the spinal level. 24
KCC2 has been implicated in various neuropathic pain. Downregulated KCC2 function induces abnormal Cl− intrusion, which results in impaired strength of GABA-A receptor mediated inhibition. 25 KCC2 enhancer could repair the above abnormal Cl− gradient and hence enhance GABA function. 26 Due to absence of GABA agonist in this study, KCC2 enhancer CLP257 itself dose-dependently counteracted the hyperalgesia state at 24 h after remifentanil exposure.
The activity of KCC2 was inevitable for the GABA to have hyperpolarizing function.27,28 KCC2 activator was demonstrated to be both a KCC2 activator and a GABAAR-positive allosteric modulator. 27 It was proposed that the functional and structural coupling of KCC2 to GABA receptor produced the comodulational effect. 29 Our present study suggested that KCC2 was downregulated after the remifentanil exposure. The KCC2 enhancer (CLP257) facilitated the anti-hyperalgesia effect of GABAAR agonist in inhibiting RIH. With absence of KCC2 enhancer, GABAAR agonist has limited inhibitory potential to ameliorate RIH.
Limitations: Our findings indicate that the RIH depressed the GABA release, followed by depressed expression of GABAAα2R. However, it was not proved that GABAAα2R downregulation is consequence of the reduced GABA release. KCC2 and GABAAα2R expression levels do not indicate altered activity in either protein. Further electrophysiological test from spinal cord slices should be performed to confirm changes in spinal nociceptive transmission. KCC2 enhancer was proved to have additive effect in potentiating the analgesic effect of GABAAR agonist. However, the mechanism of the augmentation effect, which was attributed to synergistic modulation or additive effect, still needed further investigation.
In conclusion, remifentanil exposure could inhibit the spinal release of the GABA neurotransmitter and followed by delayed inhibition of GABAAα2 receptors. Both GABAAR agonist and KCC2 enhancer were proved to dose-dependently reverse RIH after operation. Moreover, insights obtained from the study may be concluded that KCC2 enhancer potentiated anti-nociceptive effect of GABAAR agonist in reversing the pathogenesis of RIH. The mechanism of how KCC2 modulated chloride extrusion, in order to enhance GABAAR-mediated analgesia in RIH model, could be further explored in future.
Supplemental Material
sj-pdf-1-mpx-10.1177_17448069221082880 – Supplemental Material for KCC2 receptor upregulation potentiates antinociceptive effect of GABAAR agonist on remifentanil-induced hyperalgesia
Supplemental Material, sj-pdf-1-mpx-10.1177_17448069221082880 for KCC2 receptor upregulation potentiates antinociceptive effect of GABAAR agonist on remifentanil-induced hyperalgesia by Yuan Gao, Wenqiang Zhan, Yushi Jin, Xiaodan Chen, Jinxia Cai, Xiaotian Zhou, Xinyi Huang, Qimin Zhao, Weijian Wang and Jiehao Sun in Molecular Pain
Footnotes
Ethical approval
The animal study was reviewed and approved by Animal Experimentation Ethics Committee of Wenzhou Medical University (No. WYDW2019-0549).
Author Contributions
JS and WW conceived and designed the study. XC, YG, WZ, YJ, JC, XZ, XH and QZ performed the experiments. YG, WZ, YJ and JS undertook the statistical analysis and wrote the manuscript. All authors contributed to the article and approved the final version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by the Chinese National Natural Science Funding grant number 81701094, Wenzhou Municipal Science and Technology Bureau grant number 2020Y0639 and the Zhejiang Provincial Natural Science Funding grant number LY20H090008.
Data availability statement
The generated datasets are available by request directed to the corresponding author.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.