
Abstract
Neuropathic pain (NP) is a common symptom in many diseases of the somatosensory nervous system, which severely affects the patient’s quality of life. Epigenetics are heritable alterations in gene expression that do not cause permanent changes in the DNA sequence. Epigenetic modifications can affect gene expression and function and can also mediate crosstalk between genes and the environment. Increasing evidence shows that epigenetic modifications, including DNA methylation, histone modification, non-coding RNA, and RNA modification, are involved in the development and maintenance of NP. In this review, we focus on the current knowledge of epigenetic modifications in the development and maintenance of NP. Then, we illustrate different facets of epigenetic modifications that regulate gene expression and their crosstalk. Finally, we discuss the burgeoning evidence supporting the potential of emerging epigenetic therapies, which has been valuable in understanding mechanisms and offers novel and potent targets for NP therapy.
Introduction
Neuropathic pain (NP) is defined as pain caused by a lesion or disease of the somatosensory nervous system, including unpleasant feelings, such as allodynia and hyperalgesia. These symptoms severely impair the patients’ quality of life, such as sleep disturbances, anxiety, and even depressive symptoms.1,2 Furthermore, NP severely influences approximately 7%–8% of the European population and causes a tremendous medical burden worldwide. 3 Because the pathogenetic mechanisms of NP have not been sufficiently elucidated, clinical treatments are not satisfactory. Therefore, it is meaningful to explore the molecular mechanisms of NP.
Epigenetic modifications regulate gene expression without changing the DNA sequence and have a long-lasting influence on the individual when responding to environmental stimuli. The mechanisms are dynamic and reversible by repressing or activating gene expression at gene promoters. 4 A few years ago, several reviews have summarized the role of epigenetic modifications in the pathogenesis of NP.5–12 However, knowledge of the epigenetic mechanisms of NP has grown substantially in recent years. Therefore, a more comprehensive review is required to provide new insights to understand the etiology and pathophysiology of NP by summarizing the updated evidence.
In this review, we summarize the latest developments in epigenetic modifications in NP. These findings highlight epigenetic alterations in the development and maintenance of NP, including neuroinflammatory responses, ion channels, unbalanced inhibitory effects, and activation of glial cells. Moreover, we also discuss epigenetic alterations including chemical modifications to the DNA itself or to DNA-associated proteins (e.g., histones) or noncoding RNAs (ncRNAs) expression or chemical modifications to the RNA and their potential crosstalk. Finally, we suggest that specific epigenetic modifications may provide a novel therapeutic approach for treating refractory diseases.
General background of epigenetics
Waddington first put forward the term “epigenetic” in 1942, 13 it refers to heritable changes in phenotype without alteration of the DNA sequence when an organism responds to the environment.14,15 Epigenetic modifications are chemical modifications that influence the structure and function of chromatin. Chromatin comprises DNA and histone proteins and provides a scaffold for the packaging of our entire genome, including the heritable material of eukaryotic cells. As the basic critical unit of chromatin, the nucleosome has 147 base pairs of DNA wrapped around a histone octamer, which consist of two dimers each of H2A, H2B, H3, and H4. 16 Epigenetic modifications include DNA methylation, histone modifications, the regulation of non-coding RNA (ncRNA), and RNA modifications.
DNA methylation is the most widely studied epigenetic modification and refers to the covalent binding of a methyl group to the fifth carbon of a cytosine residue, known as 5-methylcytosine (5mC). This process can influence genome stability and repress gene expression. DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs), which are divided into two categories according to their function: maintenance methyltransferases (DNMT1) and de novo methyltransferases (DNMT3). The primary location of DNA methylation is cytosine-phosphate-guanine (CpG) islands, which are located in the gene promoter. 17 Increased methylation of CpG islands in the gene promoter can influence the stability of chromatin, therefore silencing gene expression. In contrast, DNA demethylation leads to the upregulation of gene expression. 18
Histone modifications are post-transcriptional modifications (PTMs) to the N-terminal histone tails of nucleosomes that regulate chromatin structure and function. These epigenetic modifications include acetylation, ubiquitination, methylation, phosphorylation, and ADP-ribosylation.19–22 Distinct PTM on histones or their combinations play a vital role in gene expression by modulating the access of transcription machinery to DNA. For example, histone acetylation leads to a loose structure that induces gene activation, while histone deacetylation results in gene repression. Histone methylation patterns and their effects on transcription are more complex than acetylation and may induce gene activation and repression depending on the residue that is methylated. 23
With regard to epigenetic modifications of gene expression, much attention has recently been devoted to ncRNAs that play critical roles in gene regulation. With the rapid development of detection technology in recent years, RNAs have also been epigenetically modified. To date, more than 100 kinds of chemical modifications of RNA have been reported.
24
Of the post-transcriptional gene regulations, the mRNA N6-methyl-adenosine (m6A) is the most studied among RNA modifications. Similar to DNA methylation and histone modifications, m6A methylation is related to the translation and degradation of RNAs
25
(Figure 1). The effects of epigenetic modifications on chromatin structure. Chromatin is primarily composed of nucleosomes, each of which consists of ∼147 base pairs of DNA wrapped around a histone octamer. The DNA sequence can be methylated at cytosine residues in a CpG context, termed DNA methylation which represses gene transcription. Histone acetylation (Ac) lead to relaxed chromatin which activate gene expression. While histone methylation facilitate activating or repressing of gene transcription depending on the location of specific lysine in histone. RNA methylation is a form of epigenetic regulation related to the translation and degradation of RNAs.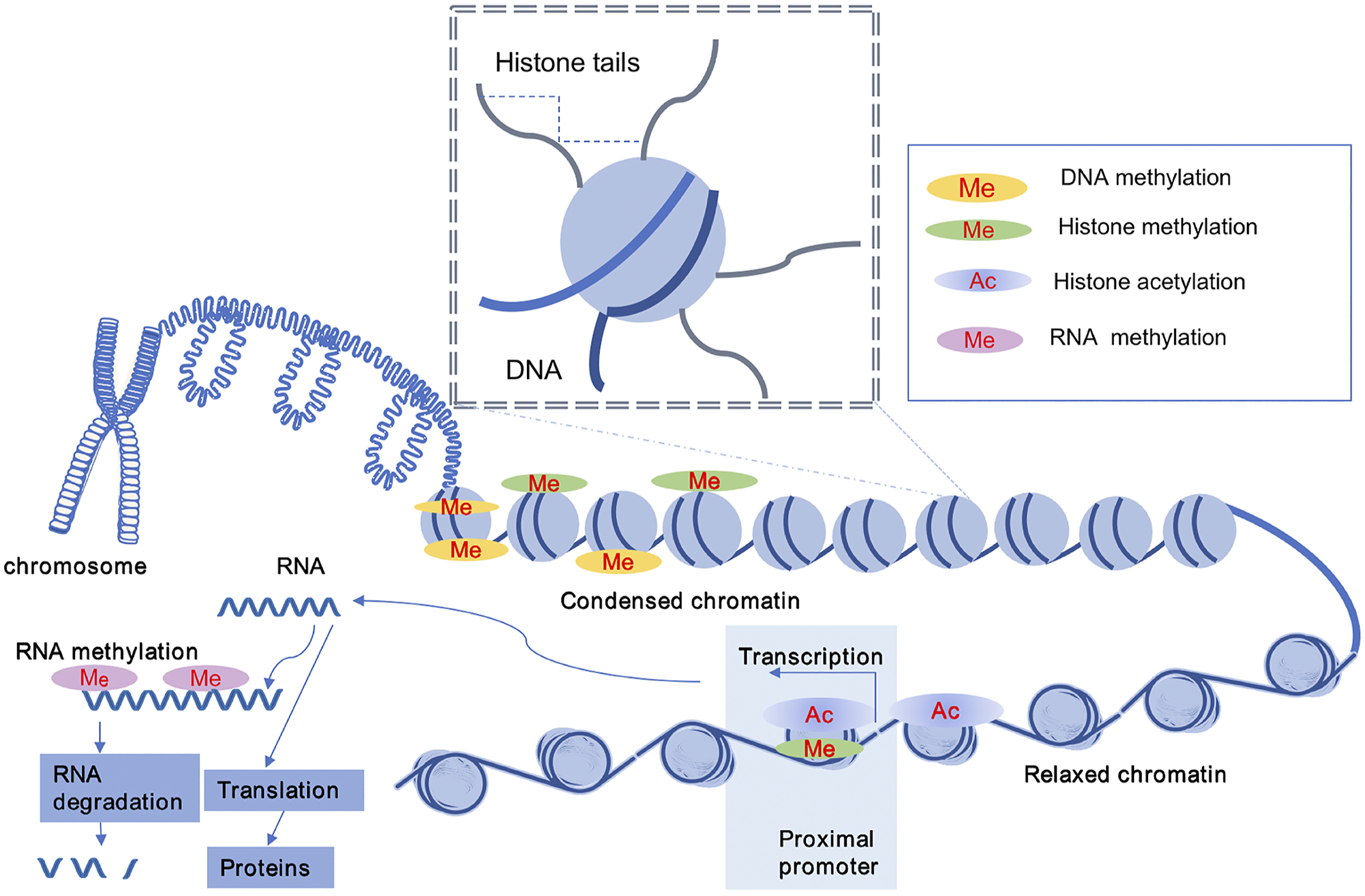
Epigenetic modifications in the mechanism of NP
It is generally accepted that NP is involved in the dysfunction of peripheral and central mechanisms, wherein inflammatory cascades trigger, neuronal firing deregulation, ion channel imbalance, glial activation, and synaptic plasticity, which lead to a lowered pain threshold.
26
For example, epigenetic modifications are involved in synaptic plasticity.
27
Here, we provide an overview of the relationship between epigenetic modification and a common mechanism of NP (Figure 2). Epigenetic modification in the mechanism of NP. After injury, immune cells, such as mast cells, macrophages and neutrophils are activated and released cytokines (e.g., IL-6, IL-1β, TNF-α) and proinflammatory mediators, which triggers inflammatory cascade. Under epigenetic modifications, the expression of ion channels are downregulated and lead to a lower pain threshold.
Neuroinflammatory response
In the development and maintenance of NP, immune cells, such as mast cells, neutrophils, macrophages, and T lymphocytes, are activated, which release numerous pro-inflammatory mediators, including cytokines, histamine, serotonin, proteases, tumor necrosis factor α (TNFα), interleukins, and chemokines.28,29 These mediators trigger an inflammatory cascade and lead to NP. 30 Furthermore, the peripheral immune response may cause ongoing activity associated with central neuroinflammation. Recently, epigenetic modifications have been found to take part in neuroinflammation during NP. Neutrophils and macrophages mediate chemokine that recruit to the injured sciatic nerve (SCN) after partial sciatic nerve ligation. The expression of macrophage inflammatory protein 2 (MIP-2)] and C-X-C chemokine receptor type 2 (CXCR2) were epigenetically up-regulated and localized on accumulated neutrophils and macrophages. 31 In the following years, the same research team also showed that epigenetic modification of CC-chemokine ligand (CCL) 2 and CCL3 mediate infiltration of immune cells to elicit NP. 32 They also indicated that vascular endothelial growth factor-A (VEGFA) is epigenetically upregulated in infiltrating macrophages and neutrophils in injured peripheral nerves. 33 However, neuroinflammation is a defense response that limits tissue damage and restores homeostasis, and anti-inflammatory cytokines also regulate these pain states. 34 There are few studies on epigenetic modification involved in anti-inflammatory cytokines in the pathogenesis of NP. Epigenetic modification of pro-inflammatory and anti-inflammatory factors adds to our understanding of the mechanism of NP.
Activation of Glial cells
As the intrinsic immune cells in the CNS, microglia serve protective functions under normal conditions. However, microglia can be activated to trigger an inflammatory cascade to promote the development of NP. 35 Epigenetic modification also participates in microglial activation. The regulator of G-protein signaling 10 (RGS10) is one of the critical anti-inflammatory regulators involved in dose-dependent suppression in response to lipopolysaccharide (LPS)-induced microglial activation. Applying HDACi trichostatin A (TSA) and HDAC1 recruitment and H3 histone deacetylation led to gene silencing. 36 Yadav et al. also showed that epigenetic modification–induced microglia activation contributes to the development of NP. They found that enhancer of zeste homolog-2 (EZH2), a subunit of the polycomb repressive complex 2, leads to gene silencing. After nerve injury, upregulation of the expression of EZH2 leads to an increase in activated microglia, and tri-methylated H3K27 (H3K27me3) is temporally associated with neuroinflammation in the spinal cord of a NP rat model. 37 Paclitaxel may cause NP, which may continue years after the therapy is stopped, 38 and is associated with microglia dysregulation. Wu et al. 39 showed that epigenetics regulates the expression of the purinergic receptor P2X4 in microglia, which causes sensitization of neurons in the dorsal horn to induce mechanical allodynia in rats. These studies provided valuable insights into the role of epigenetic modification in microglial activation associated with different models of NP.
Compared with the microglial response, the astrocytic response is delayed, but is sustained for a more extended period. 40 Astrocytes also release pro-inflammatory cytokines and chemokines to respond to harmful stimuli. There is some research showing that epigenetics mediates the activation of astrocytes in NP. CXCR3 is a chemokine receptor. Cxcr3 mRNA and protein expression are significantly epigenetically upregulated in the neurons of the spinal cord in spared nerve injury (SNI) models. In addition, the expression of chemokines, such as CXCL10, one of the ligands of CXCR3, was also increased in both neurons and astrocytes. The epigenetic modification of Cxcr3 gene causes mechanical allodynia and heat hyperalgesia. 41 In addition, Xu et al. showed that the phosphorylation of signal transducers and activators of transcription factor 3 (STAT3) and the acetylation of histone H4 in CXCL12-expressing cells are increased after microtubule-targeted agents–induced NP. The phosphorylation of the transcription factor STAT3 and the acetylation of histone H4 in CXCL12-expressing cells was increased, accompanied by mechanical allodynia. 42 Sanna et al. also found that HDAC1 is associated with the expression of c-Jun N-terminal kinase (JNK) and c-Jun in astrocytes in the spinal dorsal horn; pain symptoms are ameliorated by administration of a selective HDAC1 inhibitor (LG325). 43
These results have shown that noxious stimuli drive activation of glial cells in epigenetic modifications of NP regulation, and that these changes are associated with pain hypersensitivity under NP conditions.
Ion channel imbalance
Ion channel imbalance is usually accompanied by spontaneous ectopic firing and hyperexcitability in the dorsal root ganglion (DRG) and spinal neurons. In addition, ion channel transcriptional and translational changes in gene expression contribute to NP occurrence by increasing neuronal excitability.26,44,45 Accumulating evidence suggests that epigenetic modification involves in ion channel changes after peripheral nerve injury.
Sodium channels play a significant role in the generation and transmission of hypoesthesia and hypoalgesia. The family of sodium channels has five isoforms (Nav1.3, Nav1.6, Nav1.7, Nav1.8, and Nav1.9), all of which have been shown to regulate nociceptive responses.46–49 Epigenetic modification was found to participate in NP mechanism. Epigenetic silencing of the Nav1.8 gene in the DRG results in hypoesthesia after nerve injury. 50 It has also been reported that administration of histone deacetylase inhibitors (HDACi) can rescue the downregulation of Nav1.8 in the DRG and ameliorate negative symptoms. 51 However, a previous study indicated that the expression of Nav1.6 is decreased in the early stages of diabetic neuropathic pain (DNP) in rat models induced by streptozotocin, but increases in later periods. 52 Ding et al. 53 also reported that epigenetic upregulation of Nav1.6 gene after lumbar five ventral root transection–induced NP via TNF-α/STAT3 pathway. These studies provide promising evidence of epigenetic modification in sodium channels expressional changes under pain conditions. However, different models of NP may drive different expressional changes in the development and maintenance of NP. So, more studies are needed to verify the expressional changes in sodium channels in the long run for NP models. These could indicate some potential treatment value for NP.
Nerve injury–induced downregulation of voltage-gated potassium channel in the DRG is another critical factor for neuronal excitability and pain hypersensitivity.54,55 Uchida et al. 56 showed that nerve injury leads to gene-mediated epigenetic silencing of Kv4.3 gene through the transcriptional suppressor NRSF in the DRG. Furthermore, Laumet et al. 57 demonstrated that euchromatic histone-lysine N-methyltransferase-2 (G9a) induces the transcriptional repression of K+ channels. G9a and DNMT participate in the downregulation of voltage-gated potassium (Kv) channel subunit Kcna2 (encoding Kv1.2) in the DRG.58–61 In paclitaxel-induced NP models, decreased expression of K2p1.1 is associated with DNMT3a. 62 Considering that Kv1.2 downregulation contributes to NP genesis; Wu et al. 63 microinjected herpes simplex virus (HSV) to overexpress the Ten-eleven translocation 1 (TET1) protein to help rescue Kv1.2 expression in the ipsilateral DRG. Thus, downregulation of voltage-gated potassium channel may be an endogenous trigger in NP development and maintenance. In this sense, developing methods for reducing NP symptoms with selective epigenetic inhibitors could be a more effective approach.
Voltage-gated calcium channels (Cav) also play a crucial role in transmitting pro-nociceptive neurotransmitters, such as glutamate and substance P, to the spinothalamic tract in the occurrence of peripheral sensitization. 64 Among them, N-type, P-/Q-type, and T-type Cav are mainly expressed in C fibers and are up-regulated in inflammatory or pathological conditions, which are also involved in transmitting pain signals.65,66 Evidence has indicated that the expression of voltage-dependent T-type calcium channel 3.2 subunit was regulated by the epigenetic mechanism in pain-related regions in the DRG and dorsal horn of spinal nerve ligation (SNL) model mice. Increasing expression of the voltage-dependent T-type calcium channel 3.2 subunit directly contributes to neuropathic allodynia. 67 However, more profound researches are relatively scarce, and the validation of their functions is not powerful enough.
Transient receptor potential (TRP) channels are non-selective cation channels, and their downregulation also plays an essential role in the NP mechanism. TRPV1, TRPM8, and TRPA1 are the TRP channels that mainly involved in peripheral sensitization68–70; the TRPA1 promoter has been differentially methylated to mediate pain sensitivity. 71 Norihiko et al. also found that epigenetically increase expression of TRPA1 is associated with burning sensation symptoms in NP. 72
G protein-coupled receptor (GPCR) is the largest transmembrane receptor superfamily, which regulates ligand-gated and voltage-gated ion channels. Furthermore, they transmit extracellular signals into cells. GPCR and its ligands play an essential role in NP, mainly including gamma-aminobutyric acid (GABA) receptors, bradykinin receptors, opioid receptors, amine receptors, prostaglandin receptors, and chemokine receptors. 73 Emerging studies have shown that epigenetic modification inhibits opioid receptors in pain signal transduction during the development of NP. The analgesic effects of opioid drugs are highly unsatisfactory, in part because opioid receptors are down-regulated by epigenetic silencing of opioid receptor genes in the first-order sensory neurons of DRG, as described below58,74–76 (Figure 4). Altogether, these data indicate that epigenetic modification is a major determinant of pain response in induction and maintenance in NP.
These insights improved our understanding of epigenetic modification in the alteration of ion channel expression during NP pathophysiology. Future studies focused on the detailed mechanisms and functional roles of epigenetic modification are required to develop targeted therapies for NP.
GABA inhibitory effects
GABA plays a crucial role in the balance between excitatory and inhibitory signals in the central nervous system (CNS). The release of GABA is diminished in the spinal dorsal horn after nerve injury, contributing to the progression of NP.77,78 Glutamate decarboxylase 67 (GAD 67) is a pivotal synthetic enzyme for GABA. Moreover, GAD1 encodes the transcription of GAD 67. Epigenetic modification may participate in NP. The expression of DNMT3a, DNMT3b, and methyl-CpG binding protein 2 (MeCP2) are upregulated at the GAD1 promoter 14 days after chronic constriction injury (CCI). In contrast, MBD2 expression is decreased, leading to decreased expression of GAD 67 in the lumbar spinal cord of rat and decreased expression of GABA. 79 However, few studies have been conducted to investigate the role of epigenetic modification with GABA. Therefore, more studies are needed to better understand the underlying mechanisms between epigenetic alteration and GABA reduction.
Different epigenetic modifications in NP
Different epigenetic modifications in NP have been found in many studies. Here, we expound on the epigenetic mechanisms including DNA methylation, histone modification, ncRNAs and RNA modification.
DNA methylation
DNA methylation is a crucial epigenetic process that modifies DNA accessibility and regulates gene expression. In recent years, increasing studies have shown that aberrant DNA methylation is associated with the pain mechanism in the brain structure, as well as cortical function.
80
DNA methylation of gene promoters is generally associated with gene silencing. Upregulation of DNMT is associated with increased methylation of the MOR gene promoter in the DRG and spinal cord, leading to downregulated opioid receptor expression. This reduced expression usually leads to undesirable opioid analgesic effects in NP.76,81 In addition, DNA methylation represses gene transcription by serving as docking sites for transcription repressors, such as the family of methyl-CpG-binding domain (MBD) proteins, including MBD1-4 and MeCP2.
82
It was reported that MBD1 represses gene expression of Oprm1 [encoding mu opioid receptor (MOR)] and Kcna2 (encoding Kv1.2) in the DRG neurons by recruiting DNMT3a to these two gene promoters.
60
(Figure 3) Decreased expression of DNMTs causes DNA demethylation. Emerging studies have shown that the downregulation of DNMT3b and decreased binding of DNMT3b to gene promoters causes hypomethylation. In turn, the transcription factor increased the binding of the specific gene promoter to activate gene expression.
83
It was confirmed that hypomethylation leads to the increased expression of the purinergic receptor P2X ligand-gated ion channel 3 (P2X3R) plasticity in DRG neurons, which contribute to NP.
84
Jiang et al.
41
also found that chemokine receptor CXCR3 is upregulated by DNA demethylation and its interaction with CCAAT/enhancer-binding protein α (C/EBPα) aggravates NP. DNA demethylation is either a passive or indirect process in mammalian cells. It is usually mediated by ten-eleven translocation (TET) proteins, including TET1, TET2 and TET3. Accumulating evidence has shown that TET1-triggered transcriptional activation at CpG sites in the promoter contributes to neuron plasticity causing NP.
85
Furthermore, TET1-triggered demethylation possibly inhibits DNMT3a from binding to the promoter regions of the gene, exposing a transcription factor binding site, and allowing the transcription factor to bind.83,85 Changes of DNA methylation in the Oprm1 gene and Kcna2 gene promoter are reversed by TET1-mediated hydroxymethylation, with a concomitant reduction in pain relief.
63
Epigenetic modification of neuropathic pain. After nerve injured, the enzymes of epigenetic modification increased and stabilized binding to pain-related genes promoters, which epigenetically regulated the expression of pro-inflammatory neuromodulators. These changes led to neuroinflammation, the activation of glial and ion channels unbalanced, which contributed to pain hypersensitivity and allodynia.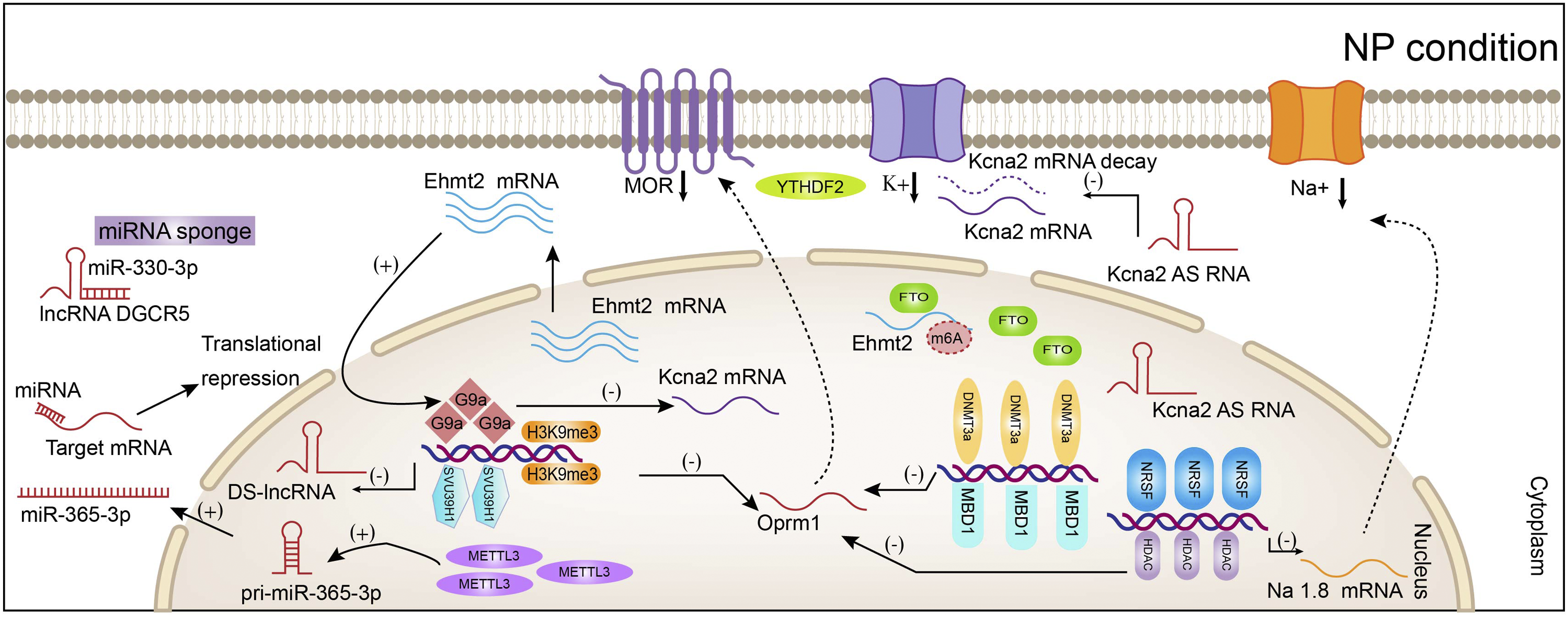
These data demonstrate that DNA methylation is critical for the development and/or maintenance of hypersensitivity to pain and this mechanism involves both MBD and reversible DNA methylation. DNA methylation is likely a potential target in neuropathic pain management.
Histone modifications
PTM of histone proteins is another significant facet of chromatin structure regulation. Histone modifications are generally associated with gene expression depending on the target residues. The most common histone modifications are acetylation and methylation.
Histone acetylation leads to gene transcriptional activation, whereas histone deacetylation causes gene repression, catalyzed by histone acetyltransferases (HATs) and reversed by the activity of histone deacetylases (HDACs). Emerging evidence has indicated that enhanced histone acetylation at promoters is associated with increased expression of pain-related genes in NP.33,86,87 The NRSF acts as a transcriptional repressor of genes. Uchida et al. found that hyperacetylation of histone H4, but not of H3, at the NRSF promoter II increases the expression of NRSF, and thus causes increased binding to the neuron-restrictive silencer the element within MOR and Nav1.8 genes that negatively influence the expression of MOR and Nav1.8. 50 (Figure 3) However, increasing histone H4 acetylation in the Scn8a promoter (encoding Nav1.6) upregulates Nav1.6 expression in DRG. 53 These results suggest that acetylation at different promoters mediates the ion channel mechanism of NP. Moreover, hyperacetylation of H3K9ac in the promoter region of inflammatory factors and chemokine ligands positively regulates their expression to induce NP.31–33 Increasing acetylation in histone H3 and H4 at brain-derived neurotrophic factor (BDNF) promoter may influence the balance of the GABA ergic and glutamatergic neurotransmissions in the spinal dorsal horn. Therefore, these dysfunctions contribute to thermal hyperalgesia and mechanical allodynia. 88 Conversely, increasing acetylation could alleviate the symptoms of NP when administrated with HDACi.36,51,89 Therefore, these studies provided valuable insights into the role of histone modification in the pathophysiological process of NP. Therefore, histone modification may be potential targets for the prevention and/or treatment of NP.
Methyl groups are added to lysine and arginine residues in histone tails in a very specific manner, including mono-, di-, and tri-methylation. Histone methylation and demethylation are catalyzed by histone methyltransferases and histone demethylases, respectively. Histone methylation plays a crucial role in the development of NP. 90 Di-methylation of Lys9 on histone H3 (H3K9me2), which is mediated by G9a, blocks K+ channel gene promoters.57, 91 In addition, another histone methyltransferase, the suppressor of variegation 3-9 homologs 1 (SUV39H1, H3K9me3), also contributes to nerve injury-induced nociceptive hypersensitivity by inhibiting MOR expression in the DRG. 92 Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine regulating neuropathic hypersensitivity by interacting with and suppressing the descending dopaminergic system. Wang et al. revealed that the recruitment of G9a and SUV39H1 leads to repression of dopamine levels, which may be associated with central sensitization.93,94 Furthermore, EZH2, a histone methyltransferase, catalyzes the methylation of histone H3 on K27 (H3K27), resulting in gene silencing. Yadav et al. showed that global levels of EZH2 and H3K27me3 in the L4–5 spinal dorsal horn are upregulated in SNL models. These upregulations may play key roles in the activation of microglia and astrocytes, and the over-production of pro-inflammatory cytokines. 37 Although previous studies have discovered the role of histone H3 lysine methylation, the functional consequences of histone H4 lysine methylation are not well understood. Future studies are needed to explore histone methylation in NP.
NcRNAs
NcRNAs are a cluster of RNAs that do not encode functional proteins and were initially considered to merely regulate gene expression at the posttranscriptional level. Generally, they can be divided into housekeeping ncRNAs and regulatory ncRNAs. RNA that has a regulatory role is mainly divided into two categories based on size: short-chain ncRNAs (including siRNAs, miRNAs, and piRNAs) and long non-coding RNA (lncRNAs).95,96 Numerous studies have shown that the epigenetic modification role of miRNAs and lncRNAs regulate the expression of specific pain-related genes in NP.
MiRNAs can negatively regulate gene expression by canonical binding to their target mRNAs or directly interact with proteins. MiRNAs mainly control gene expression by binding to the 3′-untranslated regions (3' UTR), causing either mRNA degradation or inhibition of protein translation. (Figure 3) For example, Pan et al. found that CXCR4 expression was increased in spinal glial cells of mice with SNL-induced NP. MiR-23a was significantly downregulated by directly bounding to 3' UTR of CXCR4 mRNA. Moreover, overexpression of miR-23a by intrathecal injection of miR-23a mimics or lentivirus reduced spinal CXCR4 attenuated SNL-induced pain hypersensitivity. They also revealed that associated with induction of NOD-like receptor protein 3 (NLRP3) inflammasome, was inhibiting of thioredoxin-interacting protein (TXNIP) reversed pain behavior elicited by SNL, miR-23a knockdown, or CXCR4 overexpression. 97 Also, DNA methylation play epigenetic modification role by regulating the transcription levels of miRNAs. Liu et al. 98 revealed that DNMT3a-mediated epigenetic suppression of miR-214-3p promoter enhanced colony-stimulating factor-1(CSF1) expression in astrocytes to aggravate neuroinflammation and neuronal hyperexcitability in SNL model rats. These results suggested that miRNA may potentially serve as novel therapeutic avenues through epigenetic modification in treating NP. In addition, Zhang et al. showed that N6-methyladenosine (m6A) modification took part in miRNA metabolism. In CFA-induced inflammatory pain model, methyltransferase-like 3 (METTL3) regulate pri-miR-365-3p processing by manipulating the binding of microprocessor protein DiGeorge critical region 8 (DGCR8)-dependent manner. 99 This study presents a novel direction that non-coding RNAs are regulated by RNA modification of NP.
LncRNA could interact with target mRNAs and participate in transcriptional silencing through transcription factor that binds to the lncRNA gene promoter. MiRNAs can negatively regulate gene expression by canonical binding to their target mRNAs or directly interact with proteins. MiRNAs mainly control gene expression by binding to the 3′-untranslated regions (3' UTR), causing either mRNA degradation or inhibition of protein translation. (Figure 3) The expression of Kcna2 antisense RNA (Kcna2 AS RNA) was increased by activating myeloid zinc finger protein 1 (MZF1) after peripheral nerve injury. Upregulation of Kcna2 AS RNA acted as cis-encoded lncRNA and suppressed the expression of the Kcna2 gene. These changes led to downregulation of the K+ channel increasing excitability in DRG neurons and induced NP symptoms. 100 Recent work revealed that a conserved lncRNA named DRG-specifically enriched lncRNA (DS-lncRNA), was downregulated in DRG of CCI model mice. The expression of DS-lncRNA was downregulated by its transcription factor called POU domain, class 4, transcription factor 3 (Pou4f3) to DS-lncRNA gene promoter. Downregulation of DS-lncRNA increased RALY-triggered Ehmt2 mRNA and its encoding G9a protein expression and correspondingly decreased the expression opioid receptors and KV1.2 channel, leading to pain hypersensitivities. Rescuing downregulation of DS-lncRNA reverses the G9a-controlled downregulation of opioid receptors and KV1.2 channel to attenuate pain hypersensitivities. 101 (Figure 3) What’s more, lncRNA may regulate neuroinflammation to develop or maintain NP through sponging miRNAs.102–106 For instance, lncRNA DGCR5 effectively becomes a miRNA sponge for miR-330-3p to regulate PDCD4 and contributed to mechanical and thermal hyperalgesia through the upregulation of inflammation-correlated biomarkers including interleukin 6 (IL-6), TNF α, and IL-1β in the CCI model. 105 (Figure 3) The identification of pain mechanism-related miRNAs and lncRNAs are needed to explore a therapeutic potential.
However, research regarding this aspect is currently in its infancy, and the mechanisms underlying their role in NP are yet to be elucidated. Thus, future studies focused on the detailed mechanisms, and epigenetic modification roles of ncRNAs are required to develop targeted therapies for NP.
RNA modification
Several modifications, such as N6-methyladenosine (m6A), have been detected in mRNA, revealing a new perspective on epi-transcriptomics. Similar to DNA modification and histone modification, m6A is also dynamic and reversible. In molecular mechanisms, m6A affects almost all aspects of mRNA metabolism, including splicing, translation, and stability, as well as miRNA maturation, playing an essential role in a variety of physiological processes. The enzymes involved in RNA modification can be divided into three categories according to their functions. 107 Although accumulating evidence has revealed the biological functions of m6A, which include cell differentiation, immune homeostasis, obesity, and cancer, few pieces of research have focused on the function of m6A in the development and maintenance of NP.108–111
The specific role and molecular mechanism of m6A methylation modification in NP are full of complexity and uncertainty, and further research in this field is still needed in the future. For example, METTL3 is the most studied “writer” m6A methylation enzyme in various systemic diseases. However, different scholars have found that it has opposite changes and effects in the same NP model. Zhang et al. found that the level of spinal m6A modification was significantly increased in Complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain model. At the same time, the expression of methyltransferase-like 3 (METTL3) in the spinal cord was enhanced. The CFA-induced pain behaviors could be prevented and reversed in the conditional knockdown of spinal METTL3 in the spinal cord. 99 However, Pan et al. recently reported the function of METTL3 in CFA-inflammatory pain is the opposite. They found that peripheral inflammation leads to a significant decrease of METTL3 expression in the CFA-induced chronic inflammatory pain model. This decrease correlated with a loss of m6A sites in Tet1 mRNA. By knocking down or conditionally deleting spinal METTL3 elevated the levels of m6A in Tet1 mRNA and reduced the binding of YTHDF2 to Tet1 mRNA, stabilized the increased expression of Tet1 mRNA and TET1 protein in spinal cord, resulting in pain hypersensitivity. 112 These opposite findings suggested that m6A enzymes may display distinct changes in different nervous tissues from different types of pain models. Moreover, there may be some m6A enzymes waiting to be unveiled. Furthermore, recent studies have observed that METTL3 protein did not exhibit a significant change in the NP model.113,114 These data again indicate that the role and mechanism of METTL3 and other m6A methylase mediated NP are pretty complex, and more evidence is needed to clarify. Moreover, the role of other m6A methylases in NP has also been reported. Li et al. reported that peripheral nerve injury increases the protein expression of FTO in the injured DRG, which likely contributes to the mechanisms of NP. Mimicking the increased expression of FTO correlates with a loss of m6A sites in Ehmt2 mRNA resulting in upregulation of Ehmt2-encoding G9a protein in SNL and CCI models. And the upregulated expression of G9a protein led to gene silencing, resulting in NP symptoms.114,115(Figure 3) FTO-triggered removal of toll-like receptor4 (Tlr4) mRNA m6A sites may participate in nociceptive hypersensitivities in hemorrhage-induced thalamic pain model. After microinjection of type IV collagenase (Coll IV), the FTO protein levels but not mRNA were time-dependently increased in the ipsilateral thalamus. The upregulation expression of FTO increase erased m6A sites in Tlr4 mRNA in the hemorrhagic thalamus, reducing the binding of YTH domain family protein 2 (YTHDF2, “reader” of m6A-binding protein) to 3’-untranslated regions of Tlr4 mRNA, stabilized the increased expression of Tlr4 mRNA and upregulated TLR4 receptors in the thalamus, resulting in NP symptoms. Therefore, FTO-mediated methylation of m6A participates in hemorrhage-induced thalamic pain by increasing Tlr4 mRNA expression in thalamic neurons. 116
These discoveries indicate that m6A modification may play a significant role in the development of pathological pain. Until now, the findings of m6A modification have been mainly focused on chronic inflammatory pain. There are few investigations concentrating on the relationship between m6A modification and NP. More studies are needed, which will help us better understand the molecular mechanism of NP.
Crosstalk of epigenetic modifications
Crosstalk between different modifications of gene regulation has been shown to play a key role in epigenetics. It has been reported that DNA methylation and several covalent histone protein post-translational modifications have a highly interactive effect rather than simply acting under harmful stimuli.117,118 Histone modification can also positively or negatively affect other modifications.23,119–121 Endogenous DNMT3a could interact with histone methyltransferase SUV39H1 enzymes, leading to gene silence. Moreover, G9a seems to act in concert with SUV39H1.122,123 In NP models, Oprm1 gene could be regulated by distinct epigenetic mechanisms. It has been shown that DNMT3a is upregulated and induces downregulation of MOR expression after peripheral nerve injury.76,81 Zhang et al.
75
also report that SUV39H1, a histone methyltransferase, is co-expressed with MOR. In the same year, other investigators found that G9a (encoded by Ehmt2), a histone 3 at lysine 9 methyltransferase, also regulates MOR expression in the development of NP.
74
However, it is unclear whether histone methylation is also highly coordinated with DNA methylation through multiple feedback loops (Figure 4). Our understanding of the properties of this epigenome network in NP is in its infancy. RNA m6A modification and epigenetic modifications also have crosstalk in pain models. TET proteins usually mediate DNA demethylation. The m6A methylase METTL3, which is responsible for the increased m6A levels of mRNA, affects mRNA degradation and translation. Pan et al. reported that METTL3 was downregulated, leading to a loss of m6A site in Tet1 mRNA. And the expression of TET1 was upregulated, which contributed to the pain hypersensitivity symptom.
112
This investigation reveals the co-transcriptional interplay between m6A and DNA demethylation. Li et al. provided the evidence that increased expression of FTO erased m6A in euchromatic histone lysine methyltransferase 2 (Ehmt2) mRNA (encoding the histone methyltransferase G9a) and upregulated the level of G9a in DRG. Increasing expression of G9a negatively regulated the transcription of MOR in SNL models.
114
The two types of demethylations are functionally correlated with significant co-occurrences of genetic downregulation.
124
However, few studies have investigated the relationship between these epigenetic modifications and how these marks can work in a coordinated state under NP conditions. Further studies are needed to explore the crosstalk occurring among these epigenetic modifications and help us understand of epigenetic mechanisms in NP. Crosstalk between different epigenetic layers. Chromatin is typically marked by multiple modifications. Crosstalk between these epigenetic marks may work in concert in the process of NP development and progression. (a) Crosstalk enforcing in gene activation. DNA demethylation mediated by Ten-eleven translocation (TET) protein and histone acetylation lead to gene activation. (b) Crosstalk resulting in gene silencing. DNA methylation and histone methylation could lead to gene silencing, and they may have multiple feedback loops to regulate the gene transcription.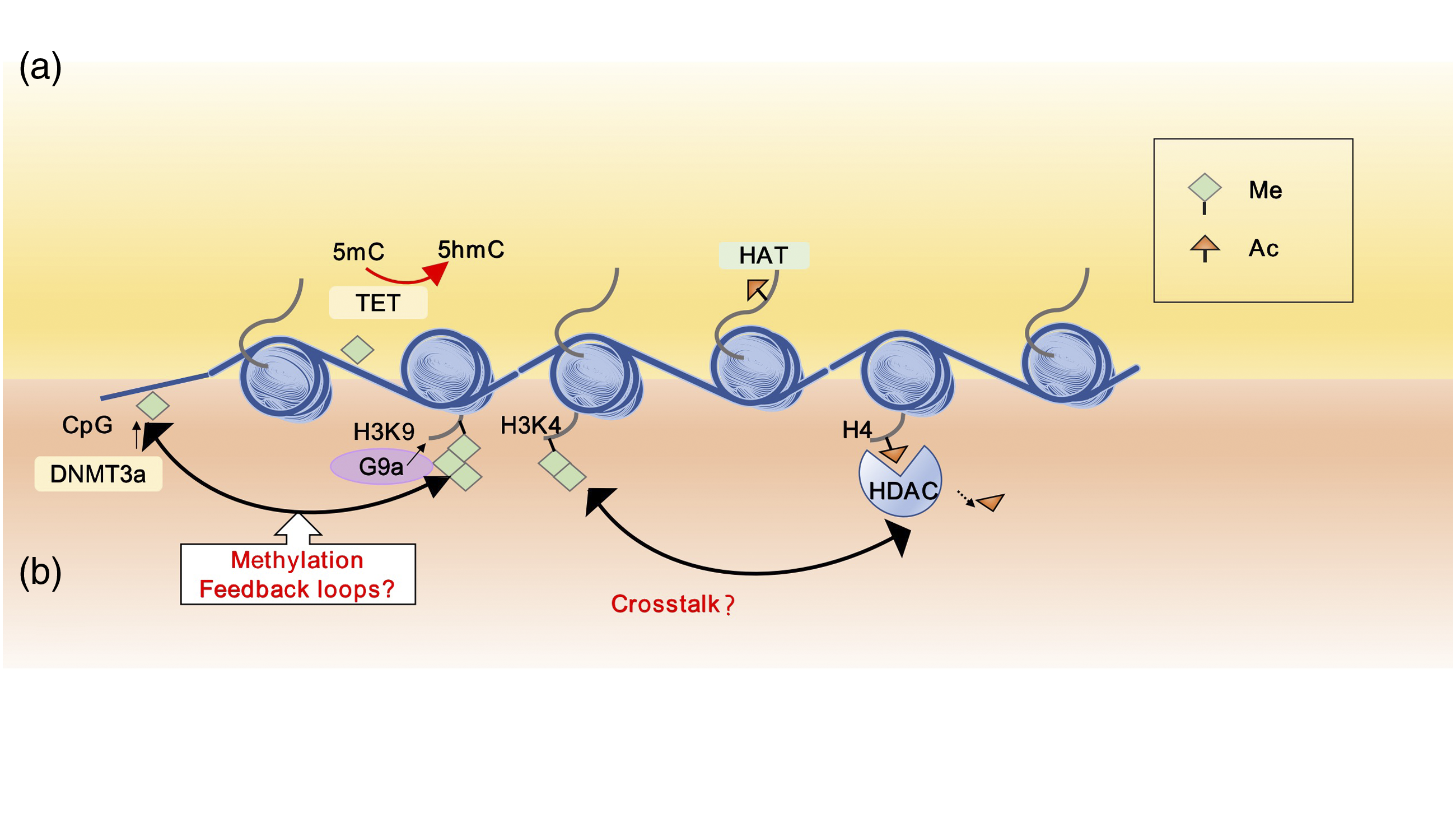
NP therapies based on epigenetic modifications
Drugs targeting the epigenetic marks in rat’s models.

Aberrant hypermethylation induced by the silencing of pain-related genes is commonly associated with the development of NP. The expression of DNMT3a is increased in the DRG and spinal cord of NP model rats with an isoform-nonspecific DNMT inhibitor N-Phthalyl-L-tryptophan (RG108) that upregulates MOR expression and attenuates thermal hyperalgesia in CCI model animals. 81 HDACi inhibit the removal of acetyl groups on histone proteins, resulting in increased histone acetylation which provides a more accessible chromatin structure with increased transcription. Increasing interest has been focused on developing of HDACi for cancer treatment, as they can modulate gene expression and the activity of numerous non-histone proteins. TSA, valproic acid (VPA), sodium phenyl butyrate, and romidepsin (LAQ-824) belong to the first generation of HDACi and are usually administered intravenously.
In contrast to the first generation of HDACi, second-generation agents, including entinostat (MS-275) and vorinostat (SAHA), can be used as oral formulations. 128 A growing number of studies have illustrated that HDACi can be significantly useful in mitigating NP symptoms in rat models (Table 1).43,51,74,89,129–131 Matsushita et al. 51 showed that administration of TSA, VPA, and SAHA significantly rescues the expression of Nav1.8, thus restoring C-fiber-related hypoesthesia. Moreover, Uchida et al. 89 found that morphine analgesia is restored when treated with HDACi such as TSA and VPA, which are accompanied by an increase in MOR expression.
Intriguingly, some dietary supplements such as S-adenosylmethionine (SAM) and folic acid (FA) which act as critical methyl donors in the CNS can attenuate SNI-induced hypomethylation and NP symptoms.132–134 Some traditional Chinese medicines have been found to ameliorate allodynia and hyperalgesia by reducing the recruitment of transcriptional factors and acetyl-histone H3/acetyl-histone H4 on pain-related genes.135,136 These discoveries have attracted renewed research attention in specific scenarios because of their potential as an analgesic adjuvant in clinical pain management.
However, most of the available epigenetic drugs are non-specific, and their long-term toxicity has not yet been tested. Therefore, epigenetics-based strategies for pain therapy are still in clinical trials. Selectivity of the epigenetic target for treating NP is essential to reduce the risk of adverse effects.
Future directions
Despite the overwhelming evidence, the pathological process of NP has not been linked to epigenetic changes. However, there are many critical questions for us to further explore epigenetic processes in NP. First, many of the experimental pain states studied are not commonly seen in clinical practice, while clinical studies in humans are lacking. Second, most of the available data on epigenetics typically use tissues such as the DRG and spinal cord in animal models. Thus, there is an urgent need to find a non-invasive alternative strategy in humans. Third, preclinical studies have focused for the most part on early time points, whereas the clinical symptoms are usually significantly prolonged. How does a primary injury result in long-lasting epigenetic marks, and how are they maintained? Could they be reversed at any time point after an injury? When does interventional therapy better reverse symptoms? However, how epigenetic marks change during the NP process is uncertain. Finally, some epigenetic modifications are preferentially expressed in cell-type–specific conditions. 137 The epigenetic inheritance of different cell types and even individual cells is unknown. Thus, future single-cell epigenetic studies will be necessary to deepen our understanding of NP.
Furthermore, neuroimaging data shows that chronic pain corresponds with changes in the amygdala, the anterior cingulate cortex, 138 the prefrontal cortex (PFC), the insular cortex, 139 and the nucleus accumbens (NAc). 140 These regions display robust interconnections with each other, and directly or indirectly modulate the activity of other pain modulating networks. Tajerian et al. found that global methylation in the prefrontal cortex and amygdala, but not the thalamus or visual cortex were decreased in SNI model after 6 months. It may reverse nerve injury-related reductions in global DNA methylation in the PFC and attenuated hypersensitivity to mechanical and cold stimuli in the environment with higher DNA methylation. 141 Further, they also reported that differential methylation in the PFC as well as DNA methylation changes in T cells was associated with peripheral nerve injury, which may be “predictors” of chronic pain. 82 Also, Topham et al. identified that pain-related genes had time point-specific differential methylation of individual genes and functional pathways in the PFC. 83 Therefore, although current studies on NP epigenetic modification mechanism mainly focus on peripheral nervous system, the above studies suggest that there may be more complex and important epigenetic modification mechanism in central mechanism, which is worthy of further exploration in the future.
Conclusion
Currently, the molecular mechanisms underlying NP and epigenetic modifications are not fully understood. However, evidence increasingly suggests that epigenetic modifications, including DNA methylation, histone modifications, and RNA modification, play critical roles in the development and maintenance of NP by regulating neuroinflammatory responses, activation of glial cells, ion channels, and unbalanced inhibitory receptors. Furthermore, communication between different epigenetic modifiers is still elusive. Fortunately, research into epigenetic modifications in NP is emerging. Epigenomics significantly extends our understanding of NP’s pathogenesis and pathophysiology and offers attractive potential for novel therapeutic approaches. With increasing knowledge of epigenetic modifications and the molecular mechanism of NP, a promising future of effective therapies for NP is not far behind.
Footnotes
Author contributions
Jun Zhou conceptualized the review. Danzhi Luo was a major contributor in writing the manuscript with the help of Xiaohong Li and Simin Tang, Fuhu Song and Wenjun Li. Guiling Xie and Jinshu Liang drew the figures and table. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grant from the National Natural Science Foundation of China (81870879) and the Natural Science Foundation of Guangdong Province(2017A030313534), Guangzhou Key Laboratory of Neuropathic Pain Mechanism at Spinal Cord Level (202102100005).