
Abstract
Research presented here sought to determine if opioid induced tolerance is linked to activity changes within the PI3Kγ-AKT-cGMP-JNK intracellular signaling pathway in spinal cord or peripheral nervous systems. Morphine or saline injections were given subcutaneously twice a day for five days (15 mg/kg) to male C57Bl/6 mice. A separate cohort of mice received spinal nerve ligation (SNL) one week prior to the start of morphine tolerance. Afterwards, spinal cord, dorsal root ganglia, and sciatic nerves were isolated for quantifying total and phosphorylated- JNK levels, cGMP, and gene expression analysis of Pik3cg, Akt1, Pten, and nNos1. This pathway was downregulated in the spinal cord with increased expression in the sciatic nerve of morphine tolerant and morphine tolerant mice after SNL. We also observed a significant increase in phosphorylated- JNK levels in the sciatic nerve of morphine tolerant mice with SNL. Pharmacological inhibition of PI3K or JNK, using thalidomide, quercetin, or SP600125, attenuated the development of morphine tolerance in mice with SNL as measured by thermal paw withdrawal. Overall, the PI3K/AKT intracellular signaling pathway is a potential target for reducing the development of morphine tolerance in the peripheral nervous system. Continued research into this pathway will contribute to the development of new analgesic drug therapies.
Neuropathic pain affects millions of patients and while the efficacy of opioid medications for neuropathic pain is debatable, opioids are still used in up to 30% of patients experiencing neuropathic pain. 1 In the nervous system, the PI3K-AKT-nNOS-cGMP intracellular signaling pathway is both implicated in both long-term pain sensitization and opioid tolerance and hyperalgesia,2,3 particularly in the spinal cord. 4 In vitro, PI3K signaling is also necessary for µ‐opioid receptor‐stimulation of c‐Jun N‐terminal kinase (JNK). 5 In rodent models, JNK inhibition in the peripheral nervous system and spinal cord were found to be involved in the development and maintenance of neuropathic pain, respectively. 6 The global loss of each of the JNK isoforms impairs morphine tolerance 7 and systemic administration of JNK inhibitors also attenuates morphine tolerance in mice. 8 Given the similarities in intracellular mechanisms involved in chronic opioid administration and pain, the research presented here sought to determine if opioid induced tolerance is linked to increased activity in the PI3Kγ-AKT-cGMP-JNK intracellular signaling pathway in either the spinal cord or the peripheral nervous system with or without underlying chronic pain conditions.
Animal protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee. Adult C57Bl/6 male mice were acquired from Charles River Laboratories (Raleigh, NC) at 5–6 weeks old. Two groups of mice were administered saline or morphine (MT, 15 mg/kg) twice a day for five days (7–8 weeks old), and a third group of mice with spinal nerve ligation (SNL) one week before chronic morphine treatment (MT + SNL 9 ). Thermal paw withdrawal latencies were performed using a modified Hargreaves radiant heat test on mice 7–8 weeks old for saline and MT and 10 weeks old for MT + SNL (TPW, 390 G, IITC, Woodland Hills, CA). 10 Vehicle (100 µL saline, subcutaneous) treated mice were used as controls for morphine tolerant mice without SNL and morphine tolerant mice after SNL (morphine 15 mg/kg, 2x day, 5 days). PI3K/AKT pathway inhibitors including quercetin (SC-206089A, 60 mg/kg, Santa Cruz Biotechnology, Dallas TX, 30 minutes before morphine), thalidomide (J60271, 100 m/kg, 100uL, Alfa Aesar, Ward Hill, MA, 15 minutes before morphine), and SP600125 (S-7979, 10 mg/kg, 100uL, LC Laboratories, Woburn, MA, 30 minutes before morphine) or vehicle (20% DMSO, 5% Tween 20, in saline, 100 µL, i.p.) controls were delivered twice per day for five days. In separate mice, spinal cord, dorsal root ganglia (DRG) and sciatic nerves were isolated from vehicle, MT, and MT+SNL mice for quantitative PCR11 or ELISA assays. mRNA was amplified using the following sense and antisense primers: Pik3cg - 5′- CAT CCA CAA AGT TCC GTC CAG-3’, 3’-GCG GAG GTT GTC CTC TCT TAG-5’; Pten -5’-TCT GCC ATC TCT CTC CTC CTT-3’, 3’- TTC TGC AGG AAA TCC CAT AGC AA-5’; nNos1 – 5’ AGC ATG ACT TCC GAG TGT GG-3’, 3’-TGC CGT CGT CGT CAT ACT TT-5’. Expression of the housekeeping gene Rn18s was compared across samples verify comparative cDNA amplification of each sample. 11 cGMP (pg/mol), phosphorylated JNK (pJNK) and total JNK (mg/ml) levels were compared across vehicle, MT, and MT+SNL treatment groups and were measured using ELISA kits according to manufacturer’s protocol. cGMP assays (5,81,021 Cayman Chemical Company, Ann-Arbor, MI) were read at 410 nmol and pJNK and total JNK (ab176662, Abcam, Cambridge, MA) assays were read at 450 nm using a BioTek Synergy 2 Plate Reader (BioTek Instruments, Inc., Winooski, VT).
Gene expression within the PI3Kγ-AKT-nNOS-cGMP intracellular signaling pathway was largely decreased in the spinal cord (Figure 1(a)) and the DRG (Figure 1(b)) and mixed in the sciatic nerves (Figure 1(c)). In particular, Pik3cg was enhanced in the ipsilateral (i.e. injured) DRG and sciatic nerves compared to vehicle treated mice. Pten and Akt1 were also enhanced in the sciatic nerves in MT+SNL tissues. cGMP was elevated in the spinal cord of MT+SNL mice (Figure 1(d)), but significantly decreased in the sciatic nerves (Figure 1(f)). pJNK was found to be elevated in the sciatic nerves of MT mice (Figure 1(f)). In behavioral studies, thalidomide, quercetin, and SP600125 treated mice had significantly increased paw withdrawal latencies compared to vehicle after five days of morphine treatment (Figure 1(g)), and a similar effect was found on the contralateral side (Figure 1(h)). Thermal paw withdrawal latencies were not significantly different after drug treatment (before morphine administration, Figure 1(i)), but were significantly increased after quercetin and SP600125 administration in the contralateral hindpaws. Interestingly, this effect was not seen after thalidomide administration (Figure 1(j)).
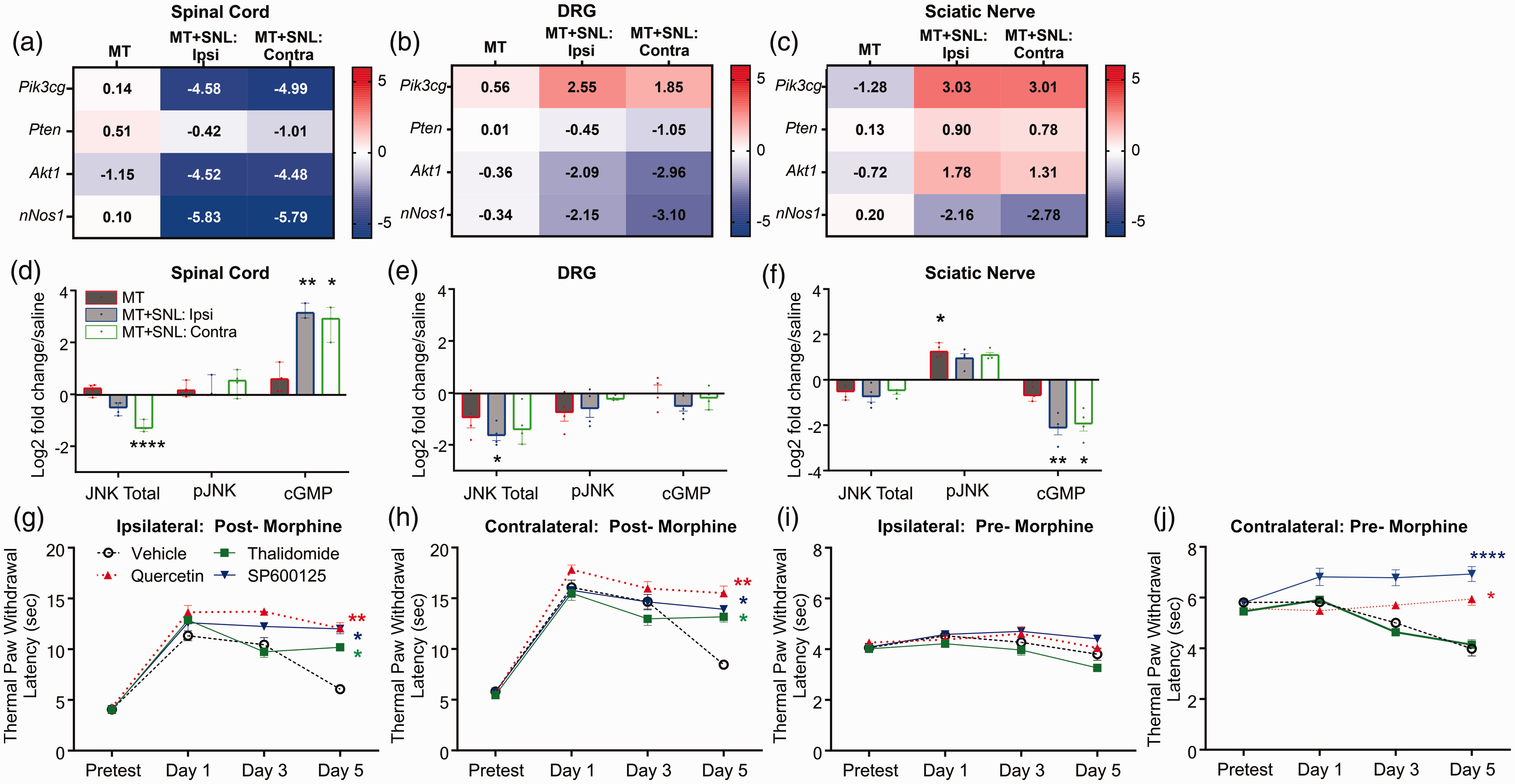
Fold change of mRNA expression in morphine-treated (MT, 15 mg/kg, x2 day, 5 days), and morphine treated mice after SNL (MT+SNL) in the spinal cord (a), dorsal root ganglia (DRG, b), and the sciatic nerve (c). mRNA counts were normalized to counts from saline treated mice and transformed as log(2). The tissues from MT+SNL mice from the injured (i.e. ipsilateral, “ipsi”) and uninjured (i.e. contralateral, “contra”) sides of mice are separated. (n = 5/group). Box color and color key show the expression level differences. The total and phosphorylated- JNK (pJNK) levels and cGMP levels are altered in in the spinal cord (d), dorsal root ganglia (e), and sciatic nerve (f) of morphine tolerant mice after spinal nerve ligation. There was a significant decrease in total JNK in the spinal cord (H(3) = 11.05, P = 0.0015), and DRG (H(3) = 8.1, p = 0.28), but an increase of pJNK in the sciatic nerves (H(3) = 8.9, p = 0.017). cGMP levels were significantly increased in the spinal cord (H(3) = 12.7, p < 0.0001), but significantly decreased in the sciatic nerves (H(3) = 13.9, p < 0.0001). Data are expressed as log2 fold change over saline-treated mice as median ± 95% CI. Kruskal-Wallis test with Dunn’s multiple comparisons test (n = 3–5/group). PI3K/AKT pathway inhibitors including quercetin (60 mg/kg), thalidomide (100 mg/kg), or SP600125 (10 mg/kg) or vehicle (20% DMSO, 5% Tween 20, in saline, 100 µL, i.p.) were administered systemically twice daily during morphine treatment. Thermal latency data were collected post-morphine treatment from the ipsilateral (g) and contralateral (h) paws of MT+SNL mice. Latency measurements were also collected pre-morphine, after vehicle or inhibitor treatment of the ipsilateral (i) or contralateral (j) hindpaws. There was a significant increase in paw latencies after morphine with thalidomide (*), quercetin (**), and SP600125 (*) treatment in the ipsilateral hindpaws (F(9,96) = 9.37, p < 0.0001, g) and contralateral hindpaws (F(9,96) = 7.69, p < 0.0001) five days after morphine treatment. A significant increase was also seen after quercetin (*) and SP600125 (****) treatment in the contralateral hindpaws before morphine treatment (F(3,32) = 49.7, p < 0.0001, j). Data expressed as mean ± SEM. Repeated measures ANOVA using a Dunnett post-hoc test (n = 5/group). Post hoc tests: *p < 0.05; **p < 0.01; ****p < 0.0001.
PI3K and its downstream target, Akt, are involved in the modulation of nociceptive information, such as neuropathic pain 12 and morphine tolerance. 13 Stimulation of downstream JNK signaling is thought to precipitate tolerance in some opioid tolerance models. 14 Our qPCR and ELISA data indicate that upregulation of JNK signaling is present in the peripheral nervous system in morphine tolerant mice after nerve injury, while the PI3Kγ and AKT are downregulated in the spinal cord. The JNK inhibitor SP600125 and quercetin, an inhibitor of PI3K and MAP kinases, were able to attenuate morphine tolerance and morphine induced hypersensitivity on the contralateral hindpaws, while thalidomide did not. This data indicates that the reversal of hypersensitivity was due to the inhibition of JNK, as the PTEN stabilizer and inhibitor of Akt phosphorylation, thalidomide, was unable to attenuate morphine induced hypersensitivity. Collectively, our data indicate that JNK-inhibition largely attenuates morphine tolerance due to effects in the peripheral nervous system. This is consistent with a previous report that intraplantar administration of the NOS-cGMP-PKG-JNK pathway inhibitors attenuate morphine tolerance in sciatic nerve injured mice. 15
Footnotes
Acknowledgments
Kayla Johnson for reading an earlier version of this manuscript and James Bjork for assistance with primer design.
Author Contributions
TO, TJ, and MM performed experiments. TO performed the statistical analysis. TO and AHK designed the experiments, discussed, and completed the final version of the paper. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding provided by the NIH to AHK (K01 DA042902), the University of Minnesota Integrated Biosciences Graduate Program to T.O., and the University of Minnesota Duluth Undergraduate Research Opportunity Program award to TJ.