
Abstract
The study reported here investigated the role of the central vascular endothelial growth factor-A (VEGF-A) pathway in the development of trigeminal neuropathic pain following nerve injury. A Sprague-Dawley rat model of trigeminal neuropathic pain was produced using malpositioned dental implants. The left mandibular second molar was extracted under anesthesia and replaced with a miniature dental implant to induce injury to the inferior alveolar nerve. The inferior alveolar nerve injury produced a significant upregulation of astrocytic VEGF-A expression in the medullary dorsal horn. The nerve injury-induced mechanical allodynia was inhibited by an intracisternal infusion of VEGF-A164 antibody. Although both VEGF-A Receptor 1 (VEGF-A R1; colocalized with the blood–brain barrier) and VEGF-A Receptor 2 (VEGF-A R2; colocalized with astrocytes) participated in the development of trigeminal neuropathic pain following nerve injury, only the intracisternal infusion of a VEGF-A R1 antibody, and not that of a VEGF-A R2 antibody, inhibited the increased blood–brain barrier permeability produced by nerve injury. Finally, we confirmed the participation of the central VEGF-A pathway in the development of trigeminal neuropathic pain by reducing VEGF-A expression using VEGF-A164 siRNA. This suppression of VEGF-A produced significant prolonged anti-allodynic effects. These results suggest that the central VEGF-A pathway plays a key role in the development of trigeminal neuropathic pain following nerve injury through two separate pathways: VEGF-A R1 and VEGF-A R2. Hence, a blockade of the central VEGF-A pathway provides a new therapeutic avenue for the treatment of trigeminal neuropathic pain.
Introduction
Vascular endothelial growth factor-A (VEGF-A) is a known key modulator of endothelial cell mitogenesis, vasculogenesis, and vascular permeability. Studies conducted in recent years have demonstrated important functional roles for VEGF-A, including the regulation of angiogenesis and lymphangiogenesis under both normal and pathological conditions.1–3 VEGF-A has neurobiological activity, including neurotrophic and neuroprotective activity, in the peripheral and central nervous systems, where it directly influences neurons, microglia, astrocytes, and Schwann cells.4,5
Recent studies have indicated that VEGF-A also participates in peripheral neuropathy resulting from nerve injury. Upregulated expression of VEGF-A was observed at the injury site or in the dorsal root ganglion following peripheral nerve injury.6,7 The perineural injection of VEGF-A inhibitor produced an inhibition of the mechanical allodynia and thermal hyperalgesia produced by partial sciatic nerve ligation. 8 Although these findings suggest that peripheral VEGF-A can modulate pain processing after peripheral injury, the role of the central VEGF-A pathway following such insults remains unclear.
Participation of VEGF in the trigeminal nerve system has been demonstrated in several previous studies. The upregulation of VEGF expression in the trigeminal ganglion was observed one month after intraperitoneal injection of streptozotocin in rats. 9 Expression of VEGF and VEGF receptors in the trigeminal ganglia has been shown to be involved in the promotion of repair after corneal nerve injury. 10 However, there has been no evidence to date of involvement of central VEGF signaling in the processing of nociceptive information arising from the orofacial area.
In the present study, we investigated the hypothesis that the central VEGF-A pathway is involved in the development of trigeminal neuropathic pain following nerve injury. We examined changes in VEGF-A expression in the medullary dorsal horn, because the medullary dorsal horn (trigeminal subnucleus caudalis) receives nociceptive information from the orofacial area, including skin, tooth pulp, and muscle. 11 We also evaluated the anti-nociceptive effects of a VEGF-A pathway blockade in a rat model of inferior alveolar nerve injury. We further examined the cellular localization of VEGF-A and its receptors in the medullary dorsal horn of these animals. To investigate the mechanisms underlying these anti-nociceptive effects, we evaluated changes in mechanical allodynia and blood–brain barrier (BBB) permeability following the blockade of VEGF-A receptors. Finally, we confirmed the participation of the central VEGF-A pathway in the development of trigeminal neuropathic pain after the reduction of VEGF-A expression via siRNA.
Materials and methods
Animals
A total of 312 male Sprague-Dawley (SD) rats weighing 200–230 g were used over all experiments. The SD rats were maintained at a constant temperature of 23 ± 1°C on a 12:12 h light–dark cycle. Food and water were freely available. All procedures involving the use of animals were approved by the Institutional Care and Use Committee of the School of Dentistry, Kyungpook National University (No. 20180108). Animal experiments were conducted in accordance with the ethical guidelines for investigations of experimental pain in conscious animals of the International Association for the Study of Pain. All experiments were performed in a blind manner.
Trigeminal neuropathic pain model
To generate a rat model of trigeminal neuropathic pain, the SD rats were anesthetized with a mixture of ketamine (40 mg/kg) and xylazine (4 mg/kg). Under anesthesia, the left mandibular second molar tooth was extracted and replaced with a mini–dental implant (diameter, 1 mm; length, 4 mm; donated by Megagen, Gyengsan, Korea) to intentionally injury the inferior alveolar nerve, as previously described.12,13 A sham group underwent surgery with no placement of a dental implant. For the control group, we used naïve rats that did not receive any operation. Only data from animals showing an inferior alveolar nerve injury caused by the malpositioned dental implants were used in the final analyses.
Intracisternal catheterization
For intracisternal administration, each individual SD rat was mounted on a stereotaxic frame and a polyethylene tube (PE 10) was implanted under anesthesia, as previously described.13–16 A hole was made in the atlanto-occipital membrane and dura using a 27-gauge needle. A polyethylene tube was inserted into the intracisternal area through the hole, and the cannula tip was placed dorsal to the obex. A polyethylene tube was then positioned subcutaneously to the skull and fixed in place by a stainless steel screw and dental acrylic resin. The animals were allowed a 72-h recovery time after surgery, because previous studies have demonstrated that this period is sufficient.17,18 Any animals showing motor dysfunction or malpositioning of the catheter after intracisternal catheterization were excluded from further analysis.
For continuous administration of drugs, a mini-osmotic pump (ALZET model 2001; Cupertino, CA) was implanted subcutaneously in the back of the neck. A polyethylene tube was then inserted through a tiny hole made in the atlantooccipital membrane. The cannula tip was connected to a mini-osmotic pump with a 1 μL/h flow rate for seven days, as described previously. 19 The mini-osmotic pump was filled with the vehicle or antibodies to be tested.
Evaluation of mechanical allodynia
Mechanical allodynia was measured as previously described.20–22 Briefly, each individual rat was placed in a customized animal cage in a darkened and noise-free room for behavioral observations. The animals were acclimated for at least 30 min. A withdrawal behavior was evaluated following 10 stimulations at a constant air-puff pressure (4-s duration and 10-s intervals) ipsilateral to the nerve injury site. We controlled the intensity and intervals of the air-puff pressure using a pico-injector (Harvard Apparatus, Holliston, MA). After inducing injury to the inferior alveolar nerve, we searched for the most sensitive area via air-puff stimulation, as previously described. 13 The most sensitive areas included the lower jaw and the mouth angle area of the facial region. We applied the air-puff stimulation to the most sensitive area through a 26-gauge metal tube of length 10 cm located 1 cm from the skin at a 90° angle. The thresholds were determined by the air-puff pressure at which each individual rat responded in 50% of the trials. The cut-off for the air-puff stimulation was 40 psi, as previously described.18,23,24 The naïve animals did not respond to a pressure lower than 40 psi.
Immunofluorescence staining
For immunofluorescence analysis, the anesthetized rats were transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Part of the caudal medulla was dissected and post-fixed in the same fixative solution at 4°C overnight. The sample was then placed in 30% sucrose solution in 0.1 M PB overnight. Transverse frozen sections (free-floating, 16 μm) were performed by a cryostat and then processed for immunofluorescence. All sections were blocked with 10% normal donkey serum in phosphate-buffered saline (PBS, pH 7.4) for 30 min at room temperature. The sections were subsequently incubated with a rabbit anti-VEGF-A (1:100; Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C conditions. After incubation, the tissue sections were washed and incubated in anti-rabbit Cy3 (1:200; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 2 h at room temperature.
For double immunofluorescence, the sections were incubated overnight at 4°C with a mixed solution of rabbit anti-VEGF-A with a mouse anti-NeuN (neuronal marker; 1:5000; Millipore, Burlington, MA), a goat anti-Iba-1 (microglial marker; 1:10,000; Abcam, Cambridge, MA), or mouse anti-GFAP (astrocytic marker; 1:10,000; Cell Signaling Technology, Danvers, MA). The sections were incubated overnight at 4°C with a rabbit anti-VEGF-A R1 (1:500; Santa Cruz) or a rabbit anti-VEGF-A R2 (1:200; Santa Cruz) mixed with a mouse anti-BBB (SMI 71; 1:1000; Biolegend, San Diego, CA) or a mouse anti-GFAP. The sections were then incubated with a mixture of anti-rabbit Cy3 (1:200; Jackson ImmunoResearch) and anti-mouse FITC (1:200; Jackson ImmunoResearch), respectively. The stained sections were observed under a fluorescence microscope
Western blotting
The rats were sacrificed under anesthesia and the ipsilateral dorsal parts of the caudal medulla were removed and frozen immediately in liquid nitrogen. The samples were subsequently sonicated with Biorupture (Cosmo Bio., Tokyo, Japan) in a lysis buffer containing a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL). We measured the concentrations of protein in the samples using a fluorometer (Invitrogen). Total proteins (30 μg) were separated on a 4–12% or 10% NuPAGE Novex Bis-Tris gel (Invitrogen) and transferred onto a nitrocellulose membrane (Invitrogen). The membranes were blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature and incubated with rabbit anti-VEGF-A antibody (1:2000; Santa Cruz), mouse anti-occludin antibody (1:1000; Santa Cruz), rabbit anti-zonula occludin (ZO)-1 antibody (1:5000; Thermo Scientific), or rabbit anti-claudin-5 antibody (1:10,000; Bioworld technology, Nanjing, China) at 4°C overnight. Mouse anti-GAPDH antibody (glyceraldehyde 3-phosphate dehydrogenase; 1:10,000; Santa Cruz) was used as a loading control. The blots were incubated with goat anti-rabbit or anti-mouse horseradish peroxidase conjugated IgG (1:5000; Bio-Rad, Hercules, CA) for 1 h at room temperature. The blots were developed using ECL kits (Millipore), followed by exposure to an Amersham Imager 600 (GE Healthcare, Little Chalfont, UK). The ImageJ analysis system (NIH, Bethesda, MD) was used to quantify the protein bands of interest.
Verification of changes in BBB permeability
BBB permeability was evaluated using Evans blue dye (Sigma-Aldrich, St Louis, MO) as previously described. 25 Briefly, at the conclusion of each experiment, Evans blue dye was injected (2% in saline, 4 ml/kg) into the right femoral vein of the anesthetized rats. Each rat was perfused with heparinized normal saline 30 min after the dye injection. The ipsilateral dorsal parts of the caudal medulla were removed, weighed, and stored at –20°C until analysis. On the day of the experiment, the tissues were incubated overnight in a 7:3 mixture of acetone and 0.5% sodium sulfate solution at room temperature with intermittent shaking. After incubation, the samples were centrifuged at 13,000 r/min for 10 min, and the supernatant was separated. The amount of dye present in the samples was analyzed by spectrophotometric measurement of the absorbance at 620 nm.
Intracisternal administration of VEGF-A164 siRNA
An siRNA molecule against VEGF-A164 and one for a nontargeting control were designed and purchased from Ambion (Carlsbad, CA) with the following sequences: sense, 5′-CAAAGAAAGAUAGAACAAAtt-3′ and antisense, 5′-UUUGUUCUAUCUUUCUUUGgt-3′. The Catalog numbers were 4390771 for the siRNA construct directed against VEGF-A164 and 4457308 for the in vivo negative control. The siRNA solution was mixed with a complexing buffer, diluted with Invivofectamine 3.0 Reagent (Invitrogen), before being incubated for 30 min at 50°C. The VEGF-A164 siRNA or negative control solutions were administered intracisternally to the rats immediately after inducing inferior alveolar nerve injury.
Experimental protocols
Statistical analysis
The behavioral data were analyzed by repeated-measures analysis of variance (ANOVA) followed by Holm-Sidak post-hoc analysis. Data from the Western blots were analyzed by one-way ANOVA followed by Holm-Sidak post-hoc analysis. In all statistical comparisons, a P value of <0.05 was considered to be significant. All data are presented as the mean ± standard error of the mean.
Results
Changes in VEGF-A expression in the medullary dorsal horn after nerve injury
The inferior alveolar nerve injury produced by the mal-positioning of dental implants in the experimental rats significantly decreased the air-puff thresholds ipsilateral to nerve injury. This nerve injury-induced mechanical allodynia presented on POD 1 and persisted until POD 30, as previously described.12,19,28 Figure 1 shows the changes in VEGF-A expression in the medullary dorsal horn on POD 5. Immunofluorescence analysis revealed that VEGF-A immunoreactivity was expressed in the ipsilateral medullary dorsal horn, where the inferior alveolar nerve was projected. Representative immunofluorescence images revealed that inferior alveolar nerve injury induced upregulation of VEGF-A expression, whereas sham-operated rats showed only weak expression of VEGF-A in the medullary dorsal horn (Figure 1(a)). Western blot analysis confirmed that changes in VEGF-A expression occurred after inferior alveolar nerve injury in the model rats, with a significant increase on POD 1, 3, and 5 (P < .05, Figure 1(b) and (c)). No significant changes were observed in VEGF-A expression in the sham-operated rats compared with the naïve rats. Accordingly, it can be concluded that inferior alveolar nerve injury increased VEGF-A expression in the ipsilateral medullary dorsal horn.
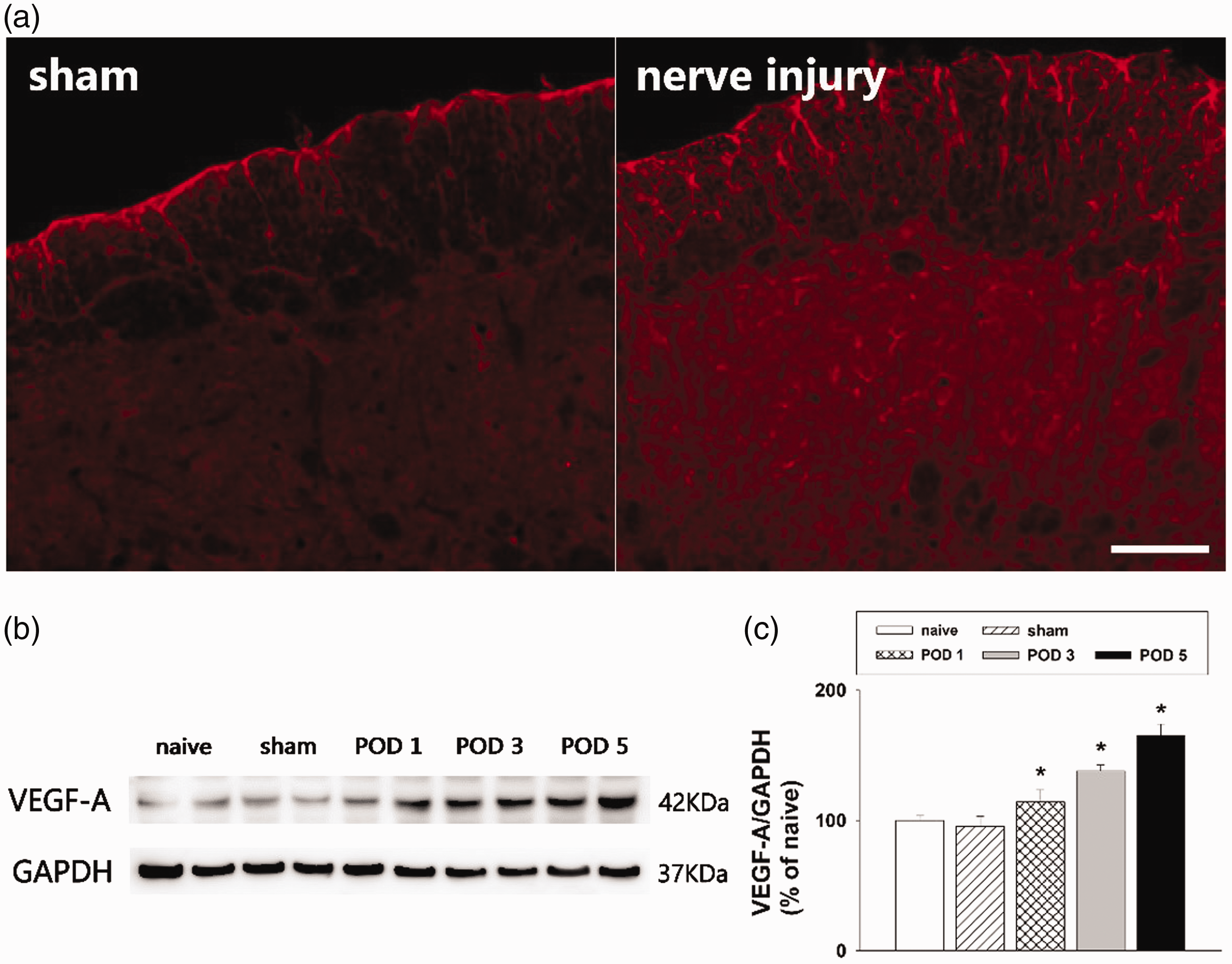
Changes in VEGF-A expression in the rat medullary dorsal horn after inferior alveolar nerve injury produced by a malpositioned dental implant. (a) Inferior alveolar nerve injury increased the number of VEGF-A immunoreactive cells in the ipsilateral medullary dorsal horn compared with the sham group. Scale bar, 100 μm. (b and c) Western blot analysis showing significantly increased VEGF-A expression on POD 1, 3, and 5 after inferior alveolar nerve injury compared with the sham group. GAPDH was used as a loading control. There were eight animals in each group. *P < .05, sham vs. nerve injury group. VEGF-A: vascular endothelial growth factor-A; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; POD: postoperative day.
Double immunofluorescence staining was performed for VEGF-A and the markers for neurons (NeuN), microglia (Iba-1), or astrocytes (GFAP), to determine its cellular localization in the medullary dorsal horn of the inferior alveolar nerve injured rats. This analysis revealed the colocalization of VEGF-A with astrocytes but not with microglia or neurons in the ipsilateral medullary dorsal horn (Figure 2).

Double immunofluorescence staining for VEGF-A (red) and either GFAP, an astrocyte marker (green); Iba-1, a microglial marker (green); or NeuN, a neuronal marker (green) on POD 5. The VEGF-A immunoreactive cells primarily colocalized with GFAP. Scale bar, 50 μm.VEGF-A: vascular endothelial growth factor-A.
Effects of VEGF-A164 antibody treatment on trigeminal mechanical allodynia
The effects of VEGF-A164 antibody treatment on mechanical allodynia are illustrated in Figure 3. The intracisternal infusion of vehicle did not affect mechanical allodynia in the animals with inferior alveolar nerve injury. However, the intracisternal infusion of VEGF-A164 antibody (250, 500 ng/day) for seven days beginning on POD 1 significantly increased the air-puff thresholds compared with the vehicle treatment (F(3, 20) = 202.3, P<.05). After treatment with 500 ng VEGF-A164 antibody, anti-allodynic effects were detectable on POD 3 and persisted up to POD 10. These anti-allodynic effects of the VEGF-A164 antibody had dissipated by POD 12 (Figure 3). The intracisternal infusion of a negative control (IgG) did not alter the mechanical allodynia in the animals with inferior alveolar nerve injury.

Effects of the intracisternal infusion of VEGF-A164 antibody for seven days on mechanical allodynia in rats with trigeminal neuropathic pain. These infusions (250 or 500 ng/day) beginning on POD 1 increased the air-puff thresholds significantly compared with the vehicle-treated group. There were six animals in each group. *P < .05, vehicle-treated vs. VEGF-A164 antibody-treated group. VEGF-A: vascular endothelial growth factor-A; POD: postoperative day.
Effects of VEGF-A receptor inhibitors on mechanical allodynia
Figure 4 illustrates the effects of VEGF-A receptor inhibitors on mechanical allodynia on POD 5. Vehicle treatment had no effects, but a single intracisternal injection of ZM306416 (20, 100 μg, a VEGF-A R1 inhibitor) produced significant anti-allodynic effects in a dose-dependent manner (F(2, 15) = 34.9, P < .05, Figure 4(a)). The administration of Vandetanib, a VEGF-A R2 inhibitor, via the same route (5, 20 μg) also produced significant anti-allodynic effects compared to the vehicle-treated group (F(2, 15) = 55.5, P < .05, Figure 4(b)). These anti-allodynic effects of the VEGF-A receptor inhibitors occurred at 90 min and had dissipated by 24 h post treatment.

Effects of a single treatment with VEGF-A receptor inhibitors on mechanical allodynia caused by inferior alveolar nerve injury. (a) The intracisternal single injection of VEGF-A R1 inhibitor (ZM306416; 20 or 100 µg) increased the air-puff thresholds. (b) The intracisternal injection of VEGF-A R2 inhibitor (Vandetanib; 5 or 20 µg) produced anti-allodynic effects compared with the vehicle-treated group. There were six animals in each group. *P < . 05, vehicle-treated vs. VEGF-A receptor inhibitors-treated group.
Colocalization of VEGF-A receptors in the medullary dorsal horn
Figure 5 illustrates the double immunofluorescence staining results for VEGF-A receptors to determine their cellular localization in the ipsilateral medullary dorsal horn on POD 5. Double immunofluorescences analyses revealed the colocalization of VEGF-A R1 with a BBB marker (SMI 71) and colocalization of VEGF-A R2 with an astrocyte marker (GFAP).

Double immunofluorescence staining for VEGF-A R1/2 (red) with either SMI 71, a BBB marker (green), or GFAP, an astrocyte marker (green), on POD 5. VEGF-A R1 colocalized with SMI 71 and VEGF-A R2 colocalized with GFAP. Scale bar, 50 μm. VEGF-A R1: VEGF-A Receptor 1; VEGF-A R2: VEGF-A Receptor 2; BBB: blood–brain barrier.
Evaluation of blood-brain barrier permeability following nerve injury
To investigate whether inferior alveolar nerve injury affects BBB permeability, the level of extravasated Evans blue dye in the ipsilateral medullary dorsal horn was examined on POD 5. No difference was found in the sham-treated group compared to the naïve group. However, inferior alveolar nerve injury significantly increased the concentration of extravasated Evans blue dye (P < .05, Figure 6(a)).

Evaluation of blood-brain barrier permeability after inferior alveolar nerve injury in rats. (a) High levels of extravasated Evans blue dye were evident on POD 5 in the inferior alveolar nerve injury group compared to the sham-treated group. There were eight animals in each group. *P < 0.05, sham vs. nerve injury group. (b and c) Western blot analysis revealed significantly decreased occludin and ZO-1 expression in the medullary dorsal horn on POD 5. Inferior alveolar nerve injury did not alter the claudin-5 expression level. GAPDH was used as a loading control. There were eight animals in each group. *P < .05, sham vs. nerve injury group. GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
We further investigated the expression of tight junction protein by using Western blot analysis to confirm changes in BBB permeability after inferior alveolar nerve injury. The levels of occludin and ZO-1 in the ipsilateral medullary dorsal horn were significantly decreased on POD 5 (P < .05, Figure 6(b) and (c)). However, inferior alveolar nerve injury did not alter the expression of claudin-5. No significant changes in the expression of these three tight-junction proteins were observed in the sham-operated rats compared to naïve rats.
Effects of blocking VEGF-A receptors on mechanical allodynia and BBB permeability
To selectively block VEGF-A receptors, VEGF-A receptor antibodies were infused intracisternally in the experimental rats for seven days beginning on POD 1. The intracisternal infusion of a negative control (IgG) did not alter mechanical allodynia in these animals with inferior alveolar nerve injury (data not shown). The intracisternal infusion of VEGF-A R1 antibody produced significant anti-allodynic effects (F(1, 14) = 319.2, P < .05). Moreover, the intracisternal infusion of VEGF-A R1 antibody decreased the concentration of extravasated Evans blue dye on POD 5 (P < .05, Figure 7(a)). The VEGF-A R2 antibody did not affect the concentration of extravasated Evan blue dye but did inhibit mechanical allodynia (F(1, 14) = 197.9, P < .05, Figure 7(b)).

Effects of VEGF-A receptor blockades on mechanical allodynia and BBB permeability in rats. (a) The intracisternal infusion of VEGF-A R1 antibody (ab, 3.5 µg/day) for seven days produced anti-allodynic effects and attenuated the increased BBB permeability produced by inferior alveolar nerve injury. There were six animals in each group. (b) The intracisternal infusion of VEGF-A R2 antibody (ab, 3.5 µg/day) for seven days also produced anti-allodynic effects but did not affect BBB permeability. There were eight animals in each group. *P < .05, vehicle- vs. VEGF-A R1/2 antibody-treated group. VEGF-A R1: VEGF-A Receptor 1; VEGF-A R2: VEGF-A Receptor 2; POD: postoperative day.
Effects of intracisternal administration of VEGF-A164 siRNA on mechanical allodynia
Figure 8 shows the effects of VEGF-A164 siRNA on neuropathic mechanical allodynia. The intracisternal injection of the negative control construct did not affect the air-puff threshold. However, VEGF-A164 siRNA produced an anti-allodynic effect in rats with trigeminal neuropathic pain at all of the tested doses (F(3, 24) = 162.7, P < .05, Figure 8(a)). A high dose (2 nmol) of VEGF-A164 siRNA produced a significantly prolonged anti-allodynic effect until eight days after a single treatment. Western blot analysis revealed that the intracisternal administration of VEGF-A164 siRNA, but not the negative control, attenuated the upregulated VEGF-A expression resulting from inferior alveolar nerve injury in the medullary dorsal horn on POD 1 and 5 (P < .05, Figure 8(b) and (c)).
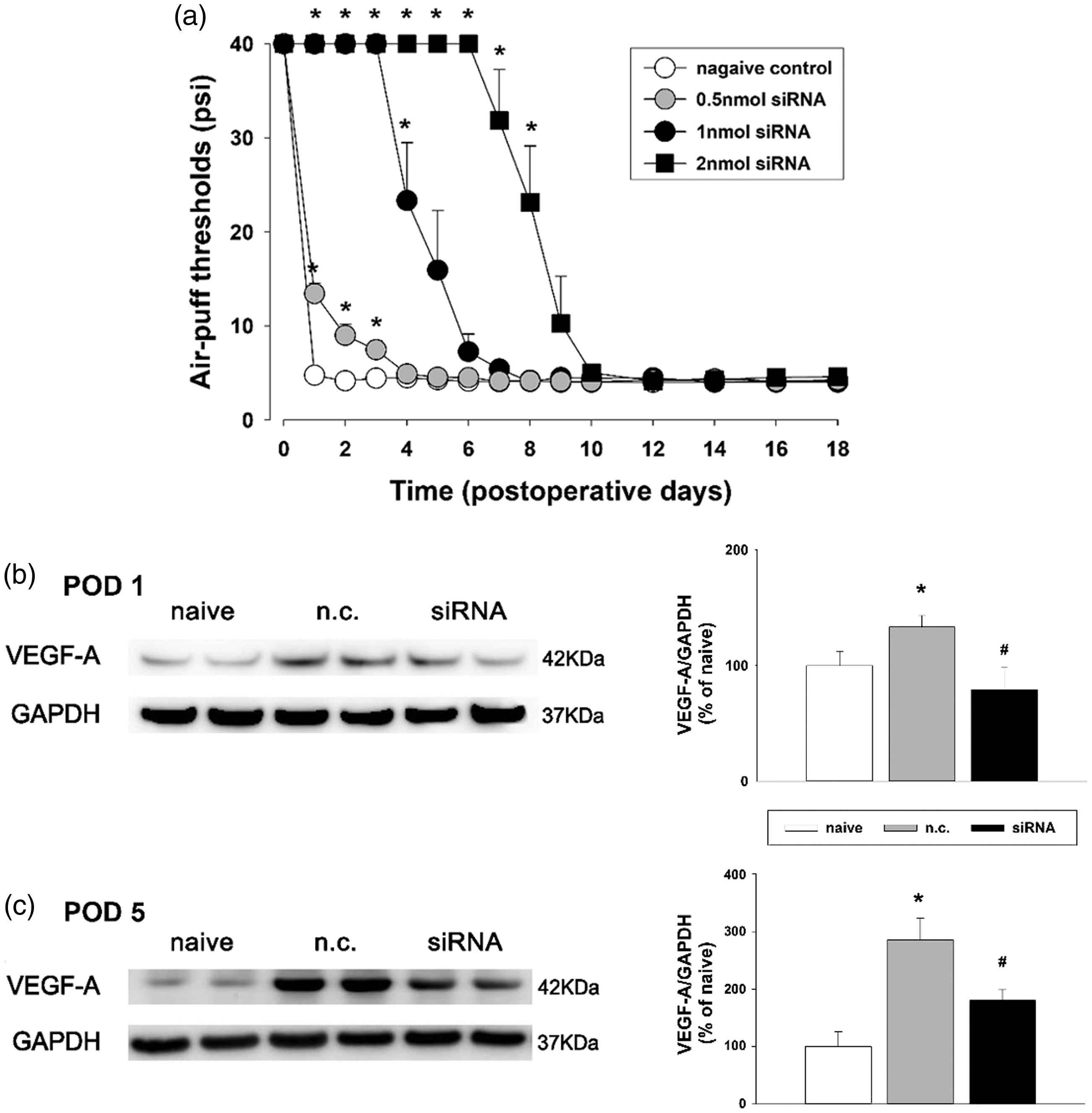
Effects of VEGF-A164 siRNA on neuropathic mechanical allodynia after inferior alveolar nerve injury in rats. (a) An intracisternal single treatment with VEGF-A164 siRNA produced significant anti-allodynic effects which persisted for eight days at the high dose (2 nmol). There were seven animals in each group. *P < . 05 vehicle- vs. VEGF-A164 siRNA-treated group. (b and c) Western blot analysis revealed that the intracisternal administration of VEGF-A164 siRNA downregulated VEGF-A expression on POD 1 and 5 in the ipsilateral medullary dorsal horn compared with the negative control (n.c.). There were eight animals in each group. *P < .05 naive vs. n.c.-treated group. #P < .05 n.c.- vs.VEGF-A164 siRNA-treated group. VEGF-A: vascular endothelial growth factor-A; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; POD: postoperative day.
Discussion
This study is the first to demonstrate that the central VEGF-A pathway plays a key role in the development of nerve injury-induced trigeminal neuropathic pain. Our findings in a rat model revealed that inferior alveolar nerve injury produced a significant upregulation of astrocytic VEGF-A expression in the medullary dorsal horn and mechanical allodynia, which were inhibited by a blockade of VEGF-A receptors. Although both VEGF-A R1 (colocalized with a BBB marker) and VEGF-A R2 (colocalized with an astrocyte marker) were found to participate in the development of trigeminal neuropathic pain following nerve injury, only the intracisternal infusion of VEGF-A R1 antibody, and not that of VEGF-A R2 antibody, inhibited the increased BBB permeability produced by nerve injury. Moreover, the downregulation of VEGF-A by VEGF-A164 siRNA produced significant, prolonged anti-allodynic effects. These results suggest that the central VEGF-A signaling pathway plays an important role in the development of trigeminal neuropathic pain after nerve injury.
Effects of a central VEGF-A pathway blockade on trigeminal neuropathic pain
Our current results demonstrated that the intracisternal infusion of VEGF-A164 antibody produced significant anti-allodynic effects. This finding is compatible with the results of previous studies, which found that a chronic constriction nerve injury increased VEGF expression in the spinal cord, 29 and that the intrathecal administration of VEGF-A antibody inhibited mechanical allodynia and thermal hyperalgesia in rats with chronic constriction injury. 7 These results indicate that the central VEGF-A pathway plays an important role in the development of neuropathic pain following nerve injury. Moreover, our findings support the contention that VEGF-A participates in the development of trigeminal neuropathic pain in the orofacial area. Inferior alveolar nerve injury in our rat model increased VEGF-A expression in the medullary dorsal horn and double immunofluorescence analysis further revealed the colocalization of VEGF-A with astrocytes in the medullary dorsal horn. These results indicate that astrocytic VEGF-A participates in the development of trigeminal neuropathic pain following nerve injury.
Changes in blood-brain barrier permeability following nerve injury
The BBB is a highly selective interface that separates the parenchyma of the central nervous system from the systemic circulation. 30 Breakdown of the BBB resulting from significant inflammation in the central nervous system leads to brain edema, excitotoxicity, and the entry of plasma proteins and inflammatory cells.6,31,32 Our investigation demonstrated that inferior alveolar nerve injury increases the concentration of extravasated Evans blue dye in the medullary dorsal horn. The evidence for this increased BBB permeability was supported by our Western blotting data showing that inferior alveolar nerve injury also decreased the expression of the tight junction proteins: occludin and ZO-1.
Notably, previous studies have demonstrated that astrocytes play an important role in BBB induction and maintenance, 33 and that VEGF-A is a potent inducer of BBB disruption.34,35 Moreover, it has been reported in a mouse model of multiple sclerosis that a blockade of VEGF-A expression in the astrocytes reduces neuropathology in inflammatory and demyelinating lesions, including decreases in BBB permeability and lymphocyte infiltration. 36 These results were shown in earlier autoimmune encephalomyelitis model. 37 These prior findings, together with our current data, suggest that astrocytic VEGF-A is one of the key drivers of BBB permeability changes following nerve injury.
Role of VEGF-A receptors in the development of trigeminal neuropathic pain
Our current analyses demonstrated that the blockade of VEGF-A R1 or R2 receptor significantly attenuated mechanical allodynia. To investigate the mechanisms underlying VEGF-A-mediated neuropathic pain following nerve injury, we evaluated changes in BBB permeability after blocking VEGF-A receptors. A blockade of VEGF-A R1 inhibited BBB permeability following nerve injury. Double immunofluorescence analysis revealed that VEGF-A R1 is colocalized with a BBB marker. These results suggest that upregulated VEGF-A expression following nerve injury produces neuropathic pain through VEGF-A R1, which consequently modulates the permeability of the BBB. The participation of VEGF-A receptor in modifying BBB permeability has been demonstrated in several previous studies, which reported that VEGF-A R1 is expressed in endothelial cells in the spinal cord following spinal cord injury in rats25,38 and in microvascular endothelial cells in human brain. 39 It has also been shown in an in vitro BBB model that selective antisense oligonucleotides directed against VEGF-A R1, but not those specific for VEGF-A R2, inhibit hypoxia-induced permeability changes. 40 These results, together with our present data, demonstrate that VEGF-A-induced hyperpermeability may be mediated by the activation of VEGF-A R1, and that the modulation of BBB permeability provides a new therapeutic strategy for chronic pain following nerve injury.
We found that the intracisternal infusion of VEGF-A R2 antibody inhibits mechanical allodynia following nerve injury in rats. However, the blockade of VEGF-A R2 did not affect BBB permeability in our animal model. Double immunofluorescence analysis revealed that VEGF-A R2 co-localizes with GFAP, an astrocyte marker. This observation suggests that VEGF-A R2 in astrocytes participates in the development of neuropathic pain, but that it does not modulate BBB permeability. These results are compatible with previous data showing that astrocytic VEGF-A protein is increased and that VEGF-A R2 expression is detectable in astrocytes of ipsilateral ischemic hemispheres. 41 Another prior study has reported that treatment with anti-recombinant VEGF-A antibody downregulates VEGF-A R2 expression and attenuates the development of neuropathic pain in rats. 42
It is well known that spinal astrocytes have a role in the development of chronic pain following nerve injury. Intrathecal injection of L-α-AA, a selective inhibitor of astrocytes, produces significant attenuation of mechanical allodynia in spinal nerve-ligated rats. 43 In the orofacial area, the activation of astrocytes in the medullary dorsal horn is associated with increases in nociceptive responses following trigeminal nerve injury. 44 Moreover, intracisternal administration of fluoroacetate, an astroglia inhibitor, reduced mechanical allodynia and thermal hyperalgesia produced by inferior alveolar nerve injury. 44 The present study demonstrated that VEGF-A R2 is colocalized with GFAP, a marker for astrocytes, and that blockade of VEGF-A R2 inhibits mechanical allodynia following inferior alveolar nerve injury in rats. These results suggest that astrocytic VEGF-A R2 plays an important role in the development of neuropathic pain following nerve injury. Additional studies are required, however, to elucidate the underlying cellular mechanisms.
Effects of VEGF-A164 siRNA on trigeminal neuropathic pain
We further confirmed the participation of VEGF-A in the development of trigeminal neuropathic pain by blocking the VEGF-A pathway using VEGF-A164 siRNA. The intracisternal administration of VEGF-A164 siRNA in our rat model attenuated the upregulated VEGF-A expression in the medullary dorsal horn produced by inferior nerve injury, while a control siRNA construct did not affect VEGF-A expression. Intracisternal treatment with VEGF-A164 siRNA produced significant, prolonged, anti-allodynic effects. These results support our hypothesis that astrocytic VEGF-A activity is a key mechanism in the development of trigeminal mechanical allodynia following nerve injury. However, the present study demonstrated that intracisternal infusion of antibody of VEGF-A164 and VEGF receptor antibodies starting on POD 1 for seven days produced anti-allodynic effects. Maximal anti-allodynic effects were observed on POD 4. These results indicate that the central VEGF pathway also plays an important role in the maintenance of neuropathic pain following nerve injury.
Limitations of the present study and clinical perspectives
In the present study, we used only male SD rats. It is well known that female showed lower pain threshold and tolerance than male related to pain. 45 Sex hormones are thought to be one of the main underlying mechanisms which explain gender differences in pain processing. 46 Hence, we used only male SD rats to exclude the effects of sex hormones on nociceptive thresholds.
The present study demonstrated that inferior alveolar nerve injury produced neuropathic mechanical allodynia ipsilateral to dental implants. However, several previous studies have shown that trigeminal nerve injury produces bilateral nociceptive behavior.12,22,47 This mirror-image neuropathic pain is created both acutely and chronically through glial and proinflammatory cytokine actions. 48 This kind of mirror-image pain is also important to the understanding of the underlying mechanisms of neuropathic pain.
For Western blotting, we used the ipsilateral dorsal parts of the caudal medulla. However, medullary structures other than medullary dorsal horn, such as solitary tract nucleus, XII nucleus, X nucleus, and dorsal column nuclei, may be included in the final analysis. This kind of unavoidable error may affect the Western blot data from the present study.
An injury to the inferior alveolar nerve is one of the most serious dental complications 49 and has a relatively high incidence in implant dentistry.50–52 This nerve damage produces complicated conditions resulted in anesthesia, paresthesia, dysesthesia, or neuropathic pain. 53 However, neuropathic pain is an intractable disease, and only 40–60% of patients with neuropathic pain achieved partial relief from pain 54 because of neuronal plasticity in the central nervous system. The present study demonstrated that a blockade of the central VEGF signaling pathway significantly inhibited trigeminal neuropathic mechanical allodynia produced by inferior alveolar nerve injury. Therefore, the modulation of the central VEGF-A pathway provides a new therapeutic target for the treatment of trigeminal neuropathic pain following nerve injury.
In conclusion, our current experiments show that inferior alveolar nerve injury produced a significant upregulation of astrocytic VEGF-A expression in the medullary dorsal horn and mechanical allodynia, which was inhibited by a VEGF-A pathway blockade. Inferior alveolar nerve injury also increased BBB permeability. Blockade of VEGF-A R1, which colocalizes with a BBB marker, produced only anti-allodynic effects. However, blockade of VEGF-A R2 (which colocalizes with an astrocyte marker) produced anti-allodynic effects and prevented the changes in BBB permeability. These results show that the central VEGF-A pathway plays a key role in the development of trigeminal neuropathic pain following nerve injury through two different pathways: VEGF-A R1 and VEGF-A R2. Hence, a blockade of the central VEGF-A pathway provides a new therapeutic avenue for the treatment of trigeminal neuropathic pain.
Footnotes
Acknowledgments
The authors thank to Megagen (Gyeongsan, Republic of Korea) for providing miniature dental implants.
Author Contributions
GW Lee, JY Son, and DK Ahn contributed to conception, design, and data analysis, drafted and critically revised the manuscript. AR Lee and JS Ju contributed to data analysis and interpretation. YC Bae contributed to conception and design and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (NRF-2017R1A5A2015391 and NRF-2018R1D1A1B07049025).