
Abstract
Elevated excitability of primary afferent neurons underlies chronic pain in patients with functional or inflammatory bowel diseases. Recent studies have established an essential role for an enhanced transient receptor potential vanilloid subtype 1 (TRPV1) signaling in mediating peripheral hyperalgesia in inflammatory conditions. Since colocalization of Toll-like receptor 4 (TLR4) and TRPV1 has been observed in primary afferents including the trigeminal sensory neurons and the dorsal root ganglion neurons, we test the hypothesis that TLR4 might regulate the expression and function of TRPV1 in primary afferent neurons in 2,4,6-trinitrobenzene sulfate (TNBS)-induced colitis using the TLR4-deficient and the wild-type C57 mice. Despite having a higher disease activity index following administration of 2,4,6-trinitrobenzene sulfate, the TLR4-deficient mice showed less inflammatory infiltration in the colon than the wild-type mice. Increased expression of TLR4 and TRPV1 as well as increased density of capsaicin-induced TRPV1 current was observed in L4–S2 dorsal root ganglion neurons of the wild-type colitis mice till two weeks post 2,4,6-trinitrobenzene sulfate treatment. In comparison, the TLR4-deficient colitis mice had lower TRPV1 expression and TRPV1 current density in dorsal root ganglion neurons with lower abdominal withdrawal response scores during noxious colonic distensions. In the wild type but not in the TLR4-deficient dorsal root ganglion neurons, acute administration of the TLR4 agonist lipopolysaccharide increased the capsaicin-evoked TRPV1 current. In addition, we found that the canonical signaling downstream of TLR4 was activated in 2,4,6-trinitrobenzene sulfate-induced colitis in the wild type but not in the TLR4-deficient mice. These results indicate that TLR4 may play a major role in regulation of TRPV1 signaling and peripheral hyperalgesia in inflammatory conditions.
Introduction
Chronic abdominal pain is one of the major manifestations in patients with various gastrointestinal (GI) disorders such as inflammatory bowel diseases (IBDs) and irritable bowel syndrome (IBS). It has been reported that significant portions of patients, including 33.3% patients with ulcerative colitis and 42%–57% patients with Crohn’s disease (CD), would remain suffering from persistent abdominal pain that is not corresponding to the inflammatory disease activity even in the quiescent phase of the disease.1–3
Numerous studies have shown that elevated excitability of the primary afferent neurons partly underlie chronic pain with accumulating evidence implicating a pivotal role of the transient receptor potential vanilloid subtype 1 (TRPV1) channel in the development of peripheral hyperalgesia. 3 TRPV1 is expressed in primary afferents and can be activated by a variety of noxious stimuli such as capsaicin (the active component of hot peppers), mustard oil, as well as noxious heat.4,5 Preclinical studies have revealed increased expression of TRPV1 in sensory neurons in rodent IBD models.6–9 In the dextran sulfate sodium (DSS)-induced colitis mice, significantly increased expression of TRPV1 was detected even after inflammation had resolved, emphasizing the contribution of TRPV1 in postinflammatory sensitization and persistent pain following acute colitis. 10 Clinical studies have also demonstrated upregulation of TRPV1 expression or increased number of TRPV1-immunoactive nerve fibers in colonic biopsies from patients with IBD or IBS, 11 highlighting TRPV1 as potential therapeutic target for chronic visceral pain.
It is now clear that active cross talk occurs between nociceptive sensory neurons and immune system, which may play important roles in pain, inflammation, and host defense. 12 Immune cells such as macrophages, mast cells, and neutrophils release numerous pro-inflammatory mediators, which may activate or sensitize visceral nociceptors. Invading pathogens or microorganisms are recognized by pattern recognition receptors expressed on innate immune cells such as macrophages. Among others, Toll-like receptors (TLRs) have been proposed to be involved in neuropathic pain. 13 As one of the most studied TLRs, TLR4 is known to recognize lipopolysaccharides (LPSs) present in cell wall of gram-negative bacteria in the gut lumen, which then interacts with its coreceptors CD14 and MD-2 to recruit the molecular myeloid differentiation marker 88 (MyD88) and downstream signaling such as nuclear factor kappa B (NF-κB) to initiate the inflammatory cascades.
Besides the well-established pro-inflammatory role, TLR4 is also implicated in modulation of pain via a direct effect on peripheral sensory neurons. TLR4, along with TLR3 and TLR7, was found to be expressed in subsets of primary sensory neurons expressing TRPV1.14,15 Paw injection of LPS was found to elicit pain-related behaviors, which were attenuated by administration of TLR4 antagonists (LPS-RS Ultrapure (LPS-RSU)).16,17 TLR4 knockout (KO) mice did not develop neuropathic pain. 18 An increased number of TLR4/TRPV1 coexpressing neurons were detected in rat and human dorsal root ganglion (DRG) neurons following paclitaxel treatment, suggesting a role for TLR4 and TRPV1 in chemotherapy-induced peripheral neuropathy (CIPN). Paclitaxel-induced behavior hypersensitivity was prevented and reversed by administration of TRPV1 antagonist. Additionally, administration of a TLR4 inhibitor (LPS-RS) prevented the increase of TRPV1-positive neurons, whereas LPS or paclitaxel activated both DRG and spinal neurons and caused an acute sensitization of TRPV1 via TLR4-mediated pathway. 19 Further investigation by the same group found paclitaxel treatment caused activation of downstream TLR4 signaling cascade in peripheral sensory neurons. 20 The above data suggest that activation of TLR4 may contribute to certain pain conditions such as CIPN and the downstream signaling may involve TRPV1. Indeed, Diogenes et al. have demonstrated that LPS may sensitize TRPV1 via TLR4 pathway in trigeminal sensory neurons, which likely underlies infection-induced oral pain. 21
Given the importance of TLR4 signaling in the GI tract, the present investigation was aimed to test the hypothesis that TLR4 may regulate the expression and function of TRPV1 in primary afferent neurons in 2,4,6-trinitrobenzene sulfate (TNBS)-induced colitis using the TLR4-deficient and the wild-type (WT) C57 mice. Our results indicate that TLR4 expressed in primary afferent neurons may play a major role in regulation of TRPV1 signaling and peripheral hyperalgesia in inflammatory conditions.
Material and methods
Animals
All animal experiments were approved by Animal Care and Use Committee of Shanghai JiaoTong University School of Medicine. Adult male mice with disrupted TLR4 gene (TLR4-KO B6.B10ScN-Tlr4lps-del/JthJ) and WT litter mates were purchased from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). Mice were housed in sterile isolator cages under specific pathogen-free conditions in animal facility of the Shanghai Jiaotong University School of Medicine.
Chemicals and reagents
Following antibodies were used: anti-TLR4 (1:1000; Abcam, Cambridge, MA), anti-TRPV1 (1:1000; Abcam), anti-NF-κB (1:1000; CST, Danvers, MA), anti-MyD88 (1:1000; CST), anti-Toll/IL-1 receptor domain-containing adapter inducing interferon-beta (TRIF) (1:500; Boster, Pleasanton, CA), anti-interleukin (IL)-6 (1:1000; Abcam), anti-IL-1β (1:500, CST), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000; Abcam) for western blotting and anti-TLR4 (1:200; Abcam) and anti-TRPV1 (1:200; Abcam) for immunofluorescence. LPSs from E. coli and capsaicin were purchased from Sigma-Aldrich (St. Louis, MO).
TNBS-induced acute colitis
Mice (WT and TLR4-deficient mice) were presensitized by epicutaneous application of 1% TNBS (5% stock solution; Sigma) in acetone and oil mixture in a volume of 100 μl. At 7 days after the presensitization, mice were anesthetized by an intraperitoneal injection of xylazine/ketamine (ketamine 100 mg/kg + xylazine 10 mg/kg), followed by intrarectal administration of 70 μl of TNBS (2.5 mg/mouse) dissolved in 50% ethanol. Control mice received 70 μl of vehicle (50% ethanol) under the same conditions. 22
Assessment of disease activity
The body weight was monitored daily for the duration of the experiment from Day 0 post TNBS or vehicle administration. Weight change was calculated as percentage change in weight compared with baseline. Stool bleeding was tested with Seroccult cards (Propper Manufacturing, Long Island City, NY) and graded as 0, no blooding; 1, trace; 2, positive; and 3, gross blooding. After the animal was euthanized through an overdose of pentobarbital and cervical dislocation, the cecum was removed and the rest of the colon was divided into proximal and distal halves. Tissue was fixed in 10% buffered formalin, paraffin embedded, sectioned, and stained with hematoxylin and eosin. Histological colonic inflammation was evaluated by an experienced pathologist who was blinded to mouse phenotype and treatment. Histological score was a combined score of inflammatory cell infiltrate (0–4) and crypt damage (0–4).23,24
Behavior test of the visceral nocifensive responses
Mice were sedated with intraperitoneal injection of xylazine/ketamine (ketamine 100 mg/kg combined with xylazine 10 mg/kg). A flexible latex balloon attached to a Tygon tubing was gently inserted into the colorectum via the anus. Mice were then put in small cubicles and allowed for 30 min to adapt. Colorectal distension (CRD) was performed by rapid inflation of the balloon to the required pressure (20, 40, 60, and 80 mmHg, respectively) for 2 s at 2 min intervals. Abdominal withdrawal responses (AWRs) to CRD were evaluated by visual inspection of the animal’s behavior and scored 0–4 according to following criteria: 0, normal behavior; 1, slight head movement without abdominal response; 2, contraction of abdominal muscles; 3, lifting of abdominal wall; and 4, body arching and lifting of pelvic structures.
Isolation of mice DRG neurons
Mice were euthanized with an overdose of pentobarbital, and L4–S2 DRGs were rapidly isolated and then transferred to cold calcium and magnesium-free phosphate-buffered saline (PBS). The isolated tissues were cut into small pieces and then incubated for 20–25 min with 0.1% collagenase and 0.025% trypsin, which were diluted by nutrient mixture F12 (DMEMDMEM-F12) at 37°C. After digestion, the DRG tissues were washed twice with DMEM-F12 containing fetal bovine serum to terminate the enzyme activity. DRG neurons were gently dissociated with polished Pasteur pipette for further patch-clamp experiments.
Patch-clamp electrophysiology
Whole-cell patch-clamp recordings of DRG neurons were carried out at room temperature (23°C–25°C) using EPC-10 amplifier and Patch Master Software (HEKA). Patch pipettes were made of borosilicate glass capillaries with a resistance of 2–5 MΩ, and the series resistance was compensated at 60% during recording. The internal solution contained (in mM): 140 CsCl, 10 N-2-hydroxyethylpiperazine-N-ethrane-sulphonicacid (HEPES), 5 ethylenebis tetraacetic acid (EGTA), 0.1 CaCl2, and 1 MgCl2, with pH adjusted to 7.2 by CsOH and osmolarity 295–300 mOsm. The external solution contained (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.4 by NaOH and osmolarity 300–310 mOsm. Signals were sampled at 10 kHz and low-pass filtered at 2.9 kHz. Capsaicin-evoked TRPV1 currents were recorded at holding membrane potential of −70 mV. Solutions were switched using a gravity-fed continuous focal perfusion system.
Quantitative real-time polymerase chain reaction
Total messenger RNA (mRNA) was extracted from T13-S2 DRG using Trisol Reagent (Invitrogen, Milano, Italy) according to manufacturer’s protocol. The extracted RNA was treated with DNase I (Invitrogen) for 20 min at 37°C to remove any genomic DNA contamination. Total RNA concentration was determined by ultraviolet spectrophotometer. Sample purity was assessed using a NanoDrop 2000 (Thermo-Fisher Scientific, Waltham, MA). Only samples with a 260/280 ratio between 1.8 and 2.0 were used for reverse transcription. First-strand cDNA was synthesized from 5 μg RNA using the SuperScript VILO cDNA Synthesis Kit (reaction conditions: 25°C for 10 min, 42°C for 2 h, and 85°C for 5 min). Real-time polymerase chain reaction (RT-PCR) was performed with primers specific for TLR4 and TRPV1. GAPDH was used as a reference gene. Primers sequences for the PCR reaction were as follows: TLR4 was TGGGTCAAGGAACAGAAGC (forward) and CAGCCAGCAATAAGTATCAGG (reverse). TRPV1 was CTGAATAACACCGTTGGGGACT (forward) and AAACTCTTGAGGGATGGTCGC (reverse). GAPDH was TGTCGGTTGTGGATCTGA (forward) and AAGGACCATGCTGTTACTTA (reverse). RT-PCR (ABI Prism 7500) was performed in a 96-well plate. Amplification conditions involved a preincubation at 95°C for 1 min followed by amplification of the target DNA for 40 cycles (95°C for 10 s and 60°C for 20 s) and fluorescence collection at 60°C. Melting curve analysis was conducted at a linear temperature transition rate of 0.5°C/s from 60°C to 95°C with continuous fluorescence acquisition. All samples were triplicated for reproducibility. The relative gene expression over GAPDH was calculated using ΔCT values, with GAPDH set at 100. 2ΔΔCT method was used to analyze target gene expression in each sample and was normalized against GAPDH.
Western blotting analysis
T13-S2 DRG from vehicle or TNBS-treated WT or TLR4-deficient mice were dissected out and put into lysed buffer (phosphatase inhibitor 1:10, proteinase inhibitor 1:100, phenylmethanesulfonyl fluoride(PMSF)1:100, and NaCl 150 mmol/l). The cell lysates were resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose filter membranes. The membranes were blocked in 25 ml blocking buffer (5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween (TBST)) for 1 h at room temperature and subsequently incubated with following primary antibodies: anti-TLR4 (1:1000; Abcam), anti-TRPV1 (1:1000; Abcam), anti-NF-κB (1:1000; CST), anti-MyD88 (1:1000; CST), anti-TRIF (1:500; Boster), anti-IL-6 (1:1000; Abcam), anti-IL-1β (1:500; CST), and anti-GAPDH (1:1000; Abcam) at 4°C overnight. After being washed with TBST buffer for three times, the blots were incubated with corresponding secondary horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies (1:5000; Abcam) for 1 h. Blots were developed using immune-western star ECL detection kits (ECL kit; Amersham Biosciences, Arlington Heights, IL). The optical densities of the immunoactive bands were analyzed by Image J software (National Institutes of Health, Bethesda, MD).
Immunohistochemistry and colocalization analysis
Mice were deeply anesthetized with sodium pentobarbital (100 mg/kg) and perfused with warm saline from ascending aorta followed by 4% cold paraformaldehyde in 0.1 M PBS (pH = 7.4). L5–L6 DRG were rapidly removed and postfixed in 4% paraformaldehyde for 6 h and cryoprotected in 30% sucrose solution overnight at 4°C. The ganglia were cut into 5 μm sections in cryostat, mounted on a gelatin-coated glass slides and then processed for immunofluorescent staining. After blocking in 5% normal serum and 0.2% Triton X-100 in PBS for 1 h at room temperature, the sections were incubated overnight at 4°C in 1% normal serum and 0.2% Triton X-100 in PBS containing primary antibodies against TLR4 (1:200; Abcam) and TRPV1 (1:200; Abcam). After washing, the sections were then incubated with fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (for TLR4, 1:1000; Abcam) or Cy5-conjugated anti-rabbit secondary antibody (for TRPV1, 1:1000; Abcam) overnight at 4°C.
Sections were visualized under confocal microscopy (Nikon A1, Tokyo, Japan). For TRPV1-positive neurons measurement in each subgroup, numbers of TRPV1-positive neurons in three DRG sections from same mouse were counted, and average value was obtained. Same procedures were performed in six mice from each group, and mean value was accessed for further analysis.
Statistical analysis
Values are presented as the mean ± standard deviation. Data were analyzed with GraphPad Prism version 6 software (GraphPad Software, San Diego, CA). Differences in disease activity index (DAI) and AWR score between groups were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. Pathology score and percentage of TRPV1 immune-positive neurons in different groups were compared using one-way ANOVA with Bonferroni’s post hoc test. Differences in the RNA and protein expression level at different time points within group were analyzed using repeated measures one-way ANOVA followed by Bonferroni’s multiple comparisons test. Differences in expression level at a given time points between two groups were compared with student’s t test. TRPV1 current density with vehicle or LPS treatment was compared using one-way ANOVA with post hoc test (Student–Newman–Keuls Method). A p value <0.05 was considered as indicating statistical significance.
Results
Characterization of TNBS-induced colitis in WT and TLR4-deficient mice
Following intrarectal administration of TNBS, both WT and TLR4-deficient mice displayed increasing disease activity characterized by gradual loss of body weight and stool bleeding. Interestingly, whereas there was no apparent difference in body weight loss between the two groups (WT 11.93 ± 0.94% vs. TLR4-KO 13.8 ± 1.12% reduction on D14, n = 6 each, p > 0.05), stool bleeding was more severe in the TLR4-deficient mice, especially during the period of D2 to D7. As a result, TNBS-treated TLR4-deficient mice had consistently higher DAI compared with the TNBS-treated WT mice (Figure 1(b)). Histological evaluation of the colonic tissue on D7 and D14 following TNBS treatment revealed tissue injury characterized by epithelial damage and neutrophilic infiltration (Figure 1(a)).25,26 Inflammatory infiltration in mucosa and submucosa was less in the TLR4-deficient colitis mice during first and second week post TNBS treatment (Figure 1(c)), whereas epithelial damage appeared to be more severe in the TLR4-deficient colitis mice than that in the WT colitis mice (Figure 1(d)). Vehicle-treated WT and TLR4-deficient mice continued to gain body weight during the two-week period (WT 20.7 ± 1.56% vs. TLR4-KO 19.8 ± 0.98% weight gain on D14, n = 6, p > 0.05).

Characterization of TNBS-induced colitis in WT and TLR4-deficient mice. (a) Photomicrographs of colonic sections from WT-TNBS, TLR4−/−-TNBS, TLR4−/−, and WT littermate controls as indicated. Scale bar = 100 μm. (b) DAI score for four subgroups during two-week period post TNBS or vehicle treatment (n = 5 per group, *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA followed by Bonferroni post hoc test). (c) Index of inflammatory infiltration for four subgroups. (d) Index of crypt epithelial damage for four subgroups (n = 5 per group, *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA followed by post hoc test). WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate.
TLR4 deficiency alleviates upregulation of TRPV1 expression in DRG neurons and visceral hypersensitivity in TNBS induces colitis
Expression of TLR4 and TRPV1 in T13-S2 DRG was evaluated on D0, D4, D7, and D14 post TNBS treatment by western blot and quantitative PCR (qPCR). In the WT mice, a concomitant incremental increase in TLR4 and TRPV1 transcription and protein expression was observed following TNBS treatment (Figure 2(a) to (c)). In the TLR4-KO mice, a similar trend of incremental increase in TRPV1 transcription and protein expression was also detected, but the increase was significantly less dramatic compared with that observed in the WT mice (Figure 2(a) to (c)). Consistent with the lower TRPV1 expression in DRG, TNBS-treated TLR4-KO mice had lower AWR scores during graded colorectal distention than TNBS-treated WT mice (Figure 2(d)). These results indicate that TLR4 signaling partly contributed to the upregulation of TRPV1 in DRG neurons and the visceral hyperalgesia in TNBS-induced colitis.

TLR4 deficiency causes reduction of TRPV1 expression in DRG and alleviation of visceral hypersensitivity in TNBS-induced colitis. (a) TLR4 and TRPV1 transcription in DRG of WT and TLR4-deficient group post TNBS treatment. (b and c) TLR4 and TRPV1 protein level in DRG of WT and TLR4-deficient mice post TNBS treatment. Differences in the expression level at different time points within group were analyzed using repeated measures one-way ANOVA followed by Bonferroni’s multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001). Differences in expression level at a given time points between two groups were compared with student’s t test (#p < 0.05; ##p < 0.01; ###p < 0.001). (d) TLR4 deficiency resulted in significant alleviation of visceral hypersensitivity in TNBS-induced colitis mice compared to control mice (*p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA followed by Bonferroni post hoc test). WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate; TRPV1: transient receptor potential vanilloid subtype 1; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; mRNA: messenger RNA.
TNBS-induced colitis increases TRPV1 current density in DRG neurons and LPS rapidly sensitizes TRPV1 through the TLR4 pathway
Previous studies have shown colocalization of TLR4 with TRPV1 in trigeminal ganglia and DRG neurons. 27 In agreement, we detected coexpression of TLR4 and TRPV1 in L5–L6 DRG neurons of the WT mice by immunohistochemistry (Figure 3). Consistent with the increased transcription and protein expression of TRPV1 in the WT mice following TNBS-induced colitis (Figure 2(a) to (c)), percentage of TRPV1-positive neurons was significantly greater in TNBS-treated than vehicle-treated mice DRG (Figure 4). Furthermore, percentage of TRPV1-positive cells was significantly lower in vehicle-treated TLR4-KO than vehicle-treated WT mice DRG. Notably, although the percentage of TRPV1-positive DRG neurons in TNBS-treated TLR4-KO mice was increased compared with the vehicle-treated TLR4-KO mice, the value was remarkably lower than that of TNBS-treated WT mice.
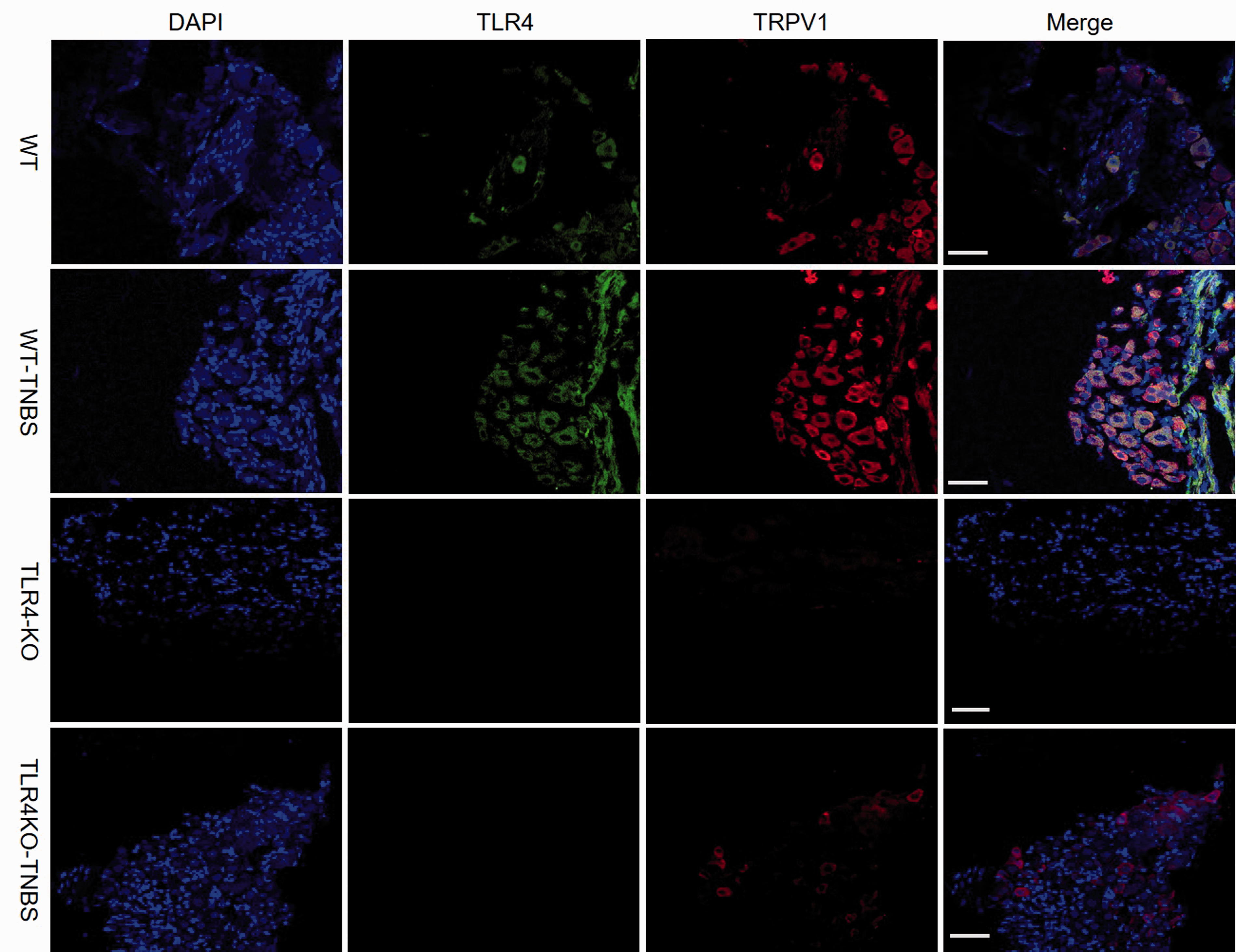
Colocalization of TRPV1 (red) and TLR4 (green) in DRG neurons. An increasing number of TRPV1-positive neurons were seen in TNBS-treated WT mice and further statistical comparisons of the percentage of TRPV1-positive neurons in different groups shown in Figure 4. Scale bar = 100 μm. WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate; TRPV1: transient receptor potential vanilloid subtype 1; DAPI: 4′,6-diamidino-2-phenylindole.
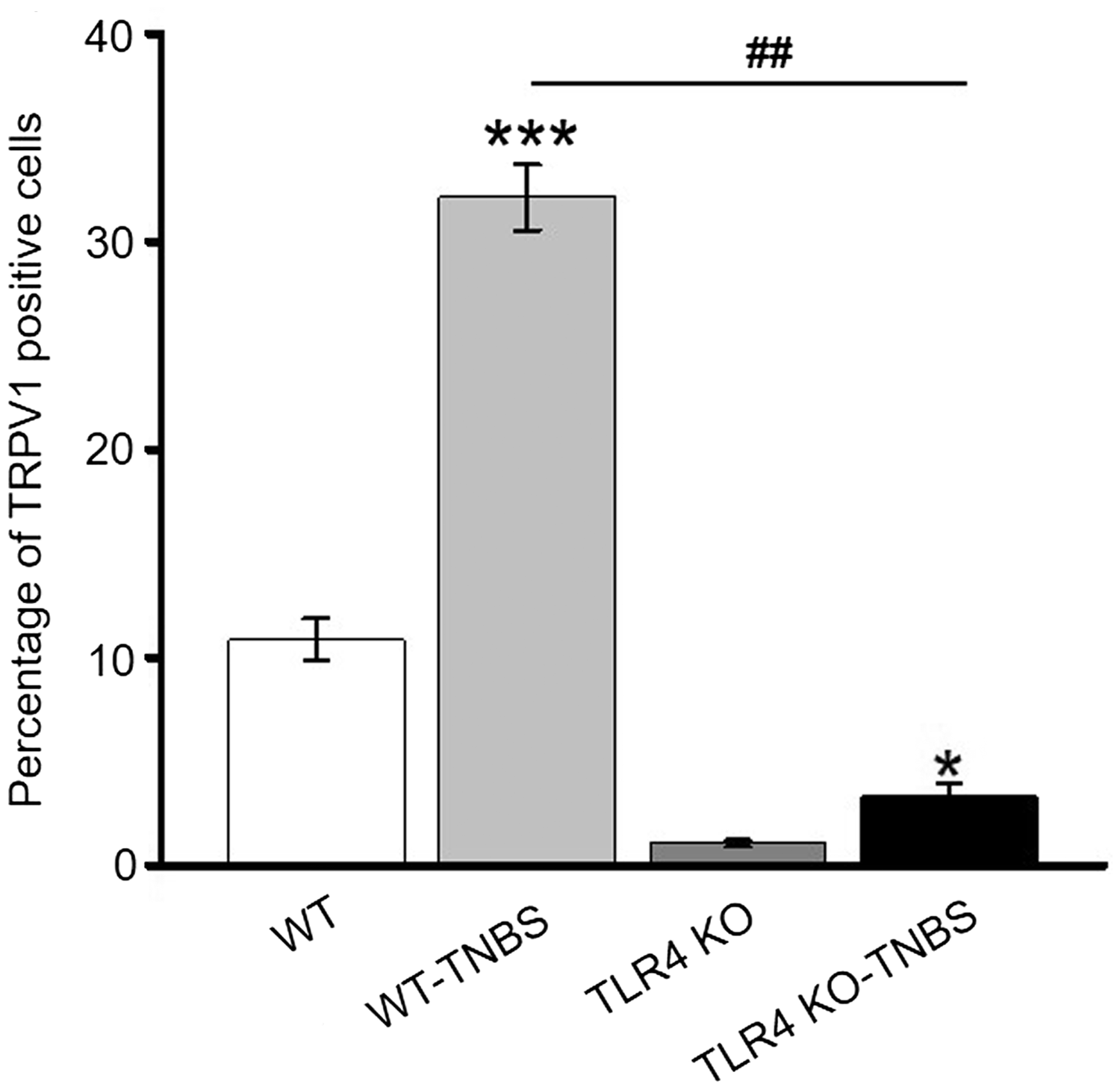
Percentage of TRPV1-positive neurons in L5–L6 DRG of four subgroups of mice. *p < 0.05; **p < 0.01, vehicle-treated WT versus TNBS-treated WT mice; ###p < 0.001, TNBS-treated TLR4-KO versus TNBS-treated WT mice (with Bonferroni’s post hoc test). WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate; TRPV1: transient receptor potential vanilloid subtype 1.
To test whether TLR4 impacts on the TRPV1 current in DRG neurons, we performed patch-clamp electrophysiology on DRG neurons from vehicle or TNBS-treated WT and TLR4-KO mice. Representative capsaicin (0.1 µM)-evoked currents were shown in Figure 5(a). TNBS-induced colitis led to a three-fold increase in capsaicin-evoked inwards current density in the WT mice (101.76 ± 14.54 pA/pF vs. 34.42 ± 8.15 pA/pF, n = 6 per group, p < 0.001, Figure 5(a) and (c)). In the TLR4-KO mice, TNBS-induced colitis also led to an increase in capsaicin-evoked TRPV1 current density in DRG neurons (Figure 5(a)). Nevertheless, TRPV1 current density in the TLR4-deficient colitis mice was significantly lower than that of the WT colitis mice (50.98 ± 4.18 pA/pF vs. 101.76 ± 14.54 pA/pF; n = 6 per group, p < 0.001, Figure 5(c)). Notably, the TLR4-deficient mice also showed significantly lower capsaicin-evoked inwards currents compared with the WT mice without colitis (10.66 ± 2.26 pA/pF vs. 34.42 ± 13.35 pA/pF; n = 6 per group, p < 0.001, Figure 5(c)).

LPS sensitizes TRPV1 through TLR4 pathway in DRG neurons. (a) Representative traces of capsaicin (0.1 µM)-evoked currents in acutely dissociated DRG neurons from four groups of mice (WT, TNBS-treated WT, TLR4-KO, and TNBS-treated TLR4-KO). Cells were treated with the vehicle for LPS for 5 min. (b) Acute treatment with LPS for 5 min increased amplitude of the capsaicin-evoked currents in WT DRG neurons. Note the scale bar is different from that of panel (a). (c) The average TRPV1 current density in the DRG neurons with vehicle or LPS treatment. *p < 0.05; **p < 0.01; ***p < 0.001(one-way ANOVA followed by Bonferroni’s multiple comparison test); #p < 0.05; ##p < 0.01; ###p < 0.001(student’s t test). WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate; LPS: lipopolysaccharide.
To further investigate whether the activation of TLR4 directly modulates TRPV1 channel activity, we observed the short-term effect of TLR4 agonist LPS on capsaicin-evoked current in DRG neurons (Figure 5(b)). Cells from the WT mice (vehicle or TNBS-treated) showed an over two-fold increase in capsaicin-evoked inwards currents after pretreatment with LPS (20 μg/ml) for 5 min, whereas the capsaicin-evoked currents of DRG neurons from the TLR4-KO mice (vehicle or TNBS-treated) were unaffected by LPS.
TNBS-induced colitis activates the canonical TLR4 downstream signaling in DRG neurons
The above data demonstrate that TLR4 colocalizes with TRPV1 in DRG neurons and that TLR4 at least partly contributes to the upregulation of TRPV1 and visceral hyperalgesia in TNBS-induced colitis. We then investigated whether TNBS-induced colitis activates the canonical TLR4 signaling in DRG neurons. Indeed, in the TNBS-treated WT mice, expression of the adaptor proteins, MyD88 and TRIF, increased rapidly till D7 and remained at high level on D14, along with elevated expression of NF-κB, IL-6, and IL-1β (Figure 6). In contrast, TLR4 deficiency resulted in no detectable expression of both adaptor proteins. Notably, however, there were incremental increases in expression of NF-κB, IL-6, and IL-1β in the DRG of TLR4-KO mice following TNBS-induced colitis. But compared with the WT colitis mice, the increases in expression of NF-κB, IL-6, and IL-1β in TLR4-KO colitis mice were less pronounced. These data collectively support the activation of the canonical TLR4 signaling might contribute to the upregulation and sensitization of TRPV1 in primary afferent neurons and visceral hyperalgesia in TNBS-induced colitis.

TNBS-induced colitis activates the canonical TLR4 signaling pathway in DRG neurons. (a) Representative western blot gel bands for adaptor proteins MyD88 and TRIF, IL-1β, IL-6, and NF-κB in the DRG from vehicle or TNBS-treated WT and TLR4-KO mice. (b) Statistic graphs showing activation of the canonical TLR4 signaling pathway in TNBS-induced colitis (n = 6 per group). *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by Bonferroni’s multiple comparisons test), #p < 0.05; ##p < 0.01; ###p < 0.001 (Student’s t test). WT: wild type; TLR4: Toll-like receptor 4; KO: knockout; TNBS: 2,4,6-trinitrobenzene sulfate; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; IL: interleukin; NF-κB: nuclear factor kappa B; TRIF: Toll/IL-1 receptor domain-containing adapter inducing interferon-beta; RU: response units.
Discussion
Chronic abdominal pain is a common clinical manifestation of functional and IBDs. Mounting evidence indicate that upregulated TRPV1 signaling in peripheral sensory neurons plays a crucial role in various chronic pain conditions, including pain arising from the GI tract.28–32 An increase in TRPV1-positive nerve fibers in colonic mucosal samples was observed to be correlated with abdominal pain scores in patients with IBS. 11 In rodent models of colitis, upregulation of TRPV1 in peripheral sensory neurons persisted after resolution of inflammation. 10 A few previous studies have also implicated TLR4 signaling in regulation of TRPV1 expression and function in sensory neurons.19,27 In the present study, we use the TLR4-deficient mice to test the hypothesis that colorectal hyperalgesia seen in TNBS-induced colitis model is associated with the activation of TLR4 signaling in DRG neurons. Consistent with the hypothesis, we found: (1) WT mice treated with TNBS displayed concomitant incremental increases in TLR4 and TRPV1 transcription and protein expression in DRG neurons; TLR4 deficiency attenuated the upregulation of TRPV1 in DRG neurons and the colorectal hyperalgesia following TNBS-induced colitis; (2) TLR4 agonist LPS rapidly increased TRPV1 current density in dissociated DRG neurons from WT but not TLR4-KO mice; (3) canonical signaling pathway downstream of TLR4 was activated in DRG neurons in WT mice treated with TNBS. Our results indicate that activation of TLR4 in DRG neurons contributes to the upregulated TRPV1 signaling and visceral hyperalgesia in TNBS-induced colitis.
TNBS-induced colitis is a common experimental model for the study of acute and postinflammatory pain. Following TNBS-induced colitis, the TLR4-deficient mice showed consistently higher DAI (combined measure of body weight loss and stool bleeding) due to more severe stool bleeding than the WT mice. Colonic epithelial damage was also more severe in the TLR4-KO than in the WT colitis mice. However, TLR4-KO mice exhibited less inflammatory infiltration in mucosa and submucosa. The more severe epithelial damage but less mucosal inflammatory infiltration in the TLR4-KO mice is perhaps explained by the dual role of TLR4 in protection of the intestinal epithelium and in inflammation. Previous studies have shown that TLR4 signaling and TLR2 signaling through the adapter molecule MyD88 were important in intestinal epithelial homeostasis and in limiting luminal bacterial translocation. Absence of TLRs signaling rendered mice more susceptible to DSS and radiation-induced injury and diminished bacteria clearance.33,34 The more severe TNBS-induced epithelial damage in the TLR4-deficient mice seen in this study was consistent with the essential role of TLR4 signaling in the maintenance of intestinal epithelial homeostasis. On the other hand, TLR4 signaling also played an crucial role in recruitment of neutrophils by upregulating expression of macrophage inflammatory protein-2 in monocytes and macrophages,35,36 which may explain the decreased inflammatory infiltration in the TLR4-KO mice in response to TNBS treatment.
Emerging evidence has already underscored TLR4 in the development of chronic pain, besides its well-known pro-inflammatory action. For example, LPS was shown to induce mechanical hyperalgesia in a dose- and time-dependent manner.37,38 Pharmacological blockade of TLR4 (e.g., TLR4 antagonist LPS-RSU) was shown to attenuate hypersensitivity in several preclinical pain models including the CIPN, cancer-induced bone pain model, the spinal cord compression injury, and the complete Freund’s adjuvant models.39–42
We found that following a single intrarectal administration of TNBS, TLR4, and TRPV1 mRNA and protein expression in T12-L6 DRGs increased continuously within the 14 days of observation in the WT mice, which underscores the role of TLR4 and TRPV1 signaling in postinflammatory visceral pain following acute colitis. This is consistent with a recent study, which showed that a single bout of DSS-induced colonic inflammation led to long lasting visceral hyperalgesia and persistent upregulation of TRPV1 up to five-week post DSS discontinuation. 10 A similar trend of increasing TRPV1 expression in DRG was detected in the TLR-deficient mice following TNBS treatment. However, TRPV1 transcription and protein expression levels were consistently lower than WT mice. In agreement with the qPCR and western blot results, we found in acutely dissociated DRG neurons that capsaicin-evoked TRPV1 current density was significantly increased in TNBS-treated compared with the vehicle-treated WT mice. However, TRPV1 current density was significantly lower in DRG neurons from TNBS-treated TLR4-deficient mice than WT colitis mice. Moreover, lower AWR score was observed in TLR4-deficient than the WT mice after TNBS-induced colitis. These results indicate that TLR4 was involved in the signaling cascade, which led to the upregulation of TRPV1 expression in DRG neurons and the colorectal hyperalgesia following colonic inflammation.
To confirm that TLR4 signaling was activated in DRG following TNBS-induced colitis, we detected the expression of adaptor proteins MyD88 and TRIF and the downstream signaling molecules, IL-1β, IL-6, and NF-κB. Indeed, expression of MyD88 and TRIF in DRG was incrementally increased along with elevated expression of NF-κB, IL-6, and IL-1β following TNBS treatment in WT mice. In TNBS-treated TLR4-deficient mice, MyD88 and TRIF were barely detectable in DRG, and the levels of IL-1β, IL-6, and NF-κB were consistently lower than those of TNBS-treated WT mice.
In addition, we found that acute treatment of the WT DRG neurons with LPS for 5 min led to an over two-fold increase in capsaicin-evoked currents. This effect was absent in DRG neurons from the TLR4-deficient mice, indicating that LPS can acutely sensitize TRPV1 via activation of TLR4. This is consistent with the findings of previous reports, which demonstrated that LPS acutely sensitizes TRPV1 function in trigeminal ganglion and DRG neurons and in HEK293 cells cotransfected with human TLR4 and TRPV1. 19
The downstream signaling engaged by TLR4 to upregulate and sensitize TRPV1 remains to be determined. However, it is well known that activation of the canonical TLR4 pathway may result in the production of pro-inflammatory cytokines such as interferon a/r, tumor necrosis factor-alpha (TNF-α), IL-1, and IL-6 and that TRPV1 signaling (expression and function) in primary afferent neurons is upregulated in inflammatory conditions. Moreover, pro-inflammatory cytokines such as TNF-αhave been shown to increase the sensitivity of TRPV1. 43 Therefore, activation (such as LPS treatment) or inactivation of TLR4 (e.g., TLR4 deficiency) likely impacts on TRPV1 signaling through an increase or a decrease in the production of pro-inflammatory cytokines.
In summary, the present study has provided evidence that activation of TLR4 in primary afferents contributes to visceral hyperalgesia in the TNBS-induced colitis model. Activation of TLR4 appears to have dual effects on TRPV1 signaling in peripheral sensory neurons, direct sensitization of TRPV1 function, and upregulation of TRPV1 transcription and protein expression.
Footnotes
Author Contributions
YWW conducted the experiments, analyzed data, and wrote the manuscript. YPW and JW conducted the in vitro experiments. QF and JZ conducted in vitro and in vivo experiments. LY conducted the experiments in core laboratory and analyzed data. WR designed and supervised the experiments and edited the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (Grant No. 81570493 and 81771963).