
Abstract
Transcription factors are proteins that modulate the transcriptional rate of target genes in the nucleus in response to extracellular or cytoplasmic signals. Activating transcription factors 2 (ATF2) and 3 (ATF3) respond to environmental signals and maintain cellular homeostasis. There is evidence that inflammation and nerve injury modulate ATF2 and ATF3 expression. However, the function of these transcription factors in pain is unknown. The purpose of this study was to investigate the contribution of ATF2 and ATF3 to nerve injury-induced neuropathic pain. L5/6 spinal nerve ligation induced tactile allodynia and thermal hyperalgesia. Moreover, nerve damage enhanced ATF2 and ATF3 protein expression in injured L5/6 dorsal root ganglia and spinal cord but not in uninjured L4 dorsal root ganglia. Nerve damage also enhanced ATF2 immunoreactivity in dorsal root ganglia and spinal cord 7 to 21 days post-injury. Repeated intrathecal post-treatment with a small-interfering RNA targeted against ATF2 (ATF2 siRNA) or anti-ATF2 antibody partially reversed tactile allodynia and thermal hyperalgesia. In contrast, ATF3 siRNA or anti-ATF3 antibody did not modify nociceptive behaviors. ATF2 immunoreactivity was found in dorsal root ganglia and spinal cord co-labeling with NeuN mainly in non-peptidergic (IB4+) but also in peptidergic (CGRP+) neurons. ATF2 was found mainly in small- and medium-sized neurons. These results suggest that ATF2, but not ATF3, found in strategic sites related to spinal nociceptive processing and participates in the maintenance of neuropathic pain in rats.
Introduction
Nerve injury induces profound changes in transcription, translation, and post-translational events in dorsal root ganglion (DRG) neurons. The cellular response to axonal damage triggers the activation of neuronal survival/death and regeneration processes. However, this cell response is maladaptive, which leads to the development and establishment of neuropathic pain.1,2
Transcription factors modulate the transcriptional rate of target genes in response to extracellular or cytoplasmic signals. Activating transcription factors (ATF) are part of the mammalian ATF/cAMP response element binding family, a large group of basic-region leucine zipper transcription factors implicated in diverse physiological functions.3,4 This family includes ATF1 (CREB), ATF2 (cAMP response element-binding protein, CRE-BP1), ATF3, ATF4, and ATF6. 4 In particular, ATF2 and ATF3 share a common ability to respond to environmental signals and maintain cellular homeostasis through the transcriptional control of stress-response and death/survival-response genes.5,6 ATF2 is constitutively expressed in many tissues, including those of the nervous system.7,8 Previous studies have reported that axotomy and complete Freund’s adjuvant injection enhance ATF2 protein expression in the DRG and spinal cord, respectively.9,10 However, the participation of ATF2 in the nociceptive processing has not been assessed.
ATF3 homodimer is a transcriptional repressor. In contrast, heterodimeric complex of ATF3 with c-Jun functions as a transcriptional activator. 4 Mounting evidence indicates that Atf3 is a stress-inducible gene. Accordingly, nerve injury, ischemia/reperfusion, chemical irritants or depletion of growth factors, among others, enhance its expression.11–14 This enhanced expression has been related to the ability of ATF3 to induce a transcriptional program favoring regeneration of damaged axons.15,16 Despite the fact that ATF3 is used as a nerve injury marker,11,14,17 the possible role of this transcription factor in neuropathic pain has not been determined. Thus, the aim of this study was to assess the participation of ATF2 and ATF3 in nerve injury-induced neuropathic pain.
Materials and methods
Animals
Since previous experiments in our conditions found no differences between male and female rats, 18 all experiments were performed on female Wistar rats (140–160 g, six weeks). Animals were obtained from our own breeding facilities and had free access to food and water before experiments. All experiments are in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985) and Guidelines on Ethical Standards for Investigation of Experimental Pain in Animals 19 and were approved by our local Ethics Committee (Protocol 042-13).
Induction of spinal nerve ligation and measurement of tactile allodynia
Rats were prepared according to a published method. 20 Briefly, animals were anesthetized with a mixture of ketamine (50 mg/kg, i.p.) and xylazine (20 mg/kg, i.p.). After surgical preparation and exposure of the dorsal vertebral column from L4 to S1, the left L5 and L6 spinal nerves were exposed and tightly ligated with 6-0 silk suture distal to the DRG. The incision was closed, and the animals were allowed to recover. In the sham group, the surgical procedure was identical to that described above, except that spinal nerves were not ligated. Animals that exhibited motor deficiency were excluded from testing (about 1%). Rats were allowed to recover from surgery for 3 to 21 days depending on the experimental group before testing pain-related behavior. Von Frey filaments (Stoelting, Wood Dale, IL, USA) were used to determine the 50% paw withdrawal threshold using the up-down method. 21 Allodynia was considered to be present when paw withdrawal thresholds were lower than 4 g.
Evaluation of thermal hyperalgesia
The paw withdrawal latency to a thermal nociceptive stimulus was evaluated as previously published. 22 Rats were placed into testing cages on a thin and clear glass plate maintained at 30°C and allowed to acclimate for about 30 min. The radiant heat source was produced by a high-intensity lamp that was activated with a timer, and the evaluator could direct it the plantar surface of the hind paw. Paw withdrawal latency was determined by a motion sensor that stopped both the radiant heat stimulus and the timer when the rat moved the leg. To prevent tissue damage, a cut-off time of 20 s was used.
Western blot analysis
Western blot analysis was used to determine the expression of ATF2 and ATF3 total protein. Naïve, sham, or ligated rats were euthanized by decapitation. The individual L4, L5, and L6 DRGs and the ipsilateral dorsal section of the lumbar enlargement (L4–L6) of the spinal cord were dissected. Immediately, tissues were frozen and stored at −70°C. Tissues from individual animals were homogenized in ice-cold lysis buffer (in mM: 150 NaCl, 50 Tris–HCl, and 1 EDTA), pH 7.4 for 2 min at 4°C. The protease inhibitors PMSF (2 mM), aprotinin (6.8 µg/ml), leupeptin (4 µg/ml), pepstatin A (4 µg/ml), and the surfactant 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) were added to the lysis buffer immediately prior to use. The homogenate was then centrifuged (Eppendorf, Germany) at 14,000 rpm for 10 min to remove cellular debris. The resultant supernatant was used to measure protein concentration by the Bradford’s method (Bio-Rad, Hercules, CA).
Fifty μg of total protein were resolved by denaturing in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis for ATF2 and ATF3 expression analysis and then transferred to polyvinylidene difluoride membranes. These were blocked in 5% non-fat milk in phosphate-buffered saline (PBS) at pH 7.4 (in mM: 137 NaCl, 2.7 KCl, 10 Na2HPO4, and 2 KH2PO4) with 0.05% Tween-20 for 1 h. After that, they were incubated overnight at 4°C in 1% non-fat dry milk/PBS containing rabbit anti-ATF2 antibody (1:500, Santa Cruz Biotechnology, Cat. # sc-187, Dallas, TX) or rabbit anti-ATF3 antibody (1:1000, Santa Cruz Biotechnology, Cat. # sc-188, Dallas, TX). Next, membranes were incubated for 1 h at room temperature in 1% non-fat milk/PBS containing the horseradish peroxidase-conjugated secondary antibody (anti-rabbit, Cat. # 111-035-003, 1:6000, Jackson Immunoresearch, West Grove, PA). Protein signal detection was achieved with the chemiluminescence system (ECL plus, Amersham, UK). The next day, blots were stripped and incubated with a monoclonal antibody against β-actin (1:10000, Cat. # GT5512, GeneTex, Irvine, CA), which was used as internal control to normalize ATF2 or ATF3 protein expression levels. Scanning of the immunoblots was performed, and the bands were quantified by densitometry using an image analysis program (Image Lab Software Version 5.2.1, Bio-Rad, Hercules, CA).
Immunohistochemistry
Independent groups of four rats each were used to determine localization of transcription factors in DRG and spinal cord. Rats were anesthetized with isoflurane and then perfused intra-aortically with 250 ml of ice-cold, 0.1 M PBS with heparin (10,000 U/L), pH 7.4, at a flow rate of 25 to 30 ml/min, followed by 4% paraformaldehyde + 12% picric acid solution in PBS. After perfusion, ipsilateral L4, L5, L6 DRGs, and spinal cord were collected and then cryoprotected for 72 h in 30% sucrose in PBS, embedded in OCT compound (Tissue-Tek; Sakura Finetek, Torrance, CA), frozen with dry ice, and stored at −70°C.
Sections were cut from frozen blocks with a cryostat (Leica CM1950, Nussloch, Germany) at a thickness of 14 µm for DRGs and 30 µm for the spinal cord. The sections were mounted onto gelatin-coated slides. Pap-pen (Super Pap-Pen, Cat. # h2802, EB Sciences, East Granby, CT) was used to draw a hydrophobic ring around the sections, and the slides were allowed to air-dry for 1 h at room temperature. Sections were first washed three times with 0.1 M PBS pH 7.4, followed by a blocking step with 3% normal donkey serum in PBS + 0.3% TX-100 for 2 h. Then, sections were incubated overnight at 4°C in 1% blocking solution containing the primary antibodies. Immunodetection of ATF2 and ATF3 in DRGs was performed using rabbit anti-ATF2 (1:500, Cat. # sc-187, Santa Cruz Biotechnology, Dallas, TX) and rabbit anti-ATF3 (1:500, Cat. # sc-188, Santa Cruz Biotechnology, Dallas, TX) antibodies, respectively. Additional markers were used to identify the immunoreactivity of neuronal nuclei, peptidergic, and non-peptidergic neurons, astrocytes, and microglia: mouse anti-NeuN antibody (1:1000, Cat. # MAB377, Merck, Darmstadt, Germany), goat anti-CGRP antibody (1:600, Cat. # 8856 N-20, Santa Cruz Biotechnology, Dallas, TX), biotinylated anti-IB4 antibody (1:600, Cat. # L2140, Sigma-Aldrich, St. Louis, MO), mouse anti-GFAP antibody (1:500, Cat. # G3893, Sigma-Aldrich, St. Louis, MO), and mouse anti-CD11b (OX-42) antibody (1:100 Cat. # MCA275G, Bio-Rad, Hercules, CA), respectively. Sections were washed in 0.1 M PBS and incubated for 3 h at room temperature with the corresponding secondary antibody conjugated to Cy3 (Cy™3 AffiniPure donkey anti-rabbit IgG [H+L], 1:600, Cat. # 711-166-152, Jackson ImmunoResearch, West Grove, PA); Cy2 (Cy™2 AffiniPure donkey anti-mouse IgG [H+L], 1:400, Cat. # 715-225-150, Jackson ImmunoResearch, West Grove, PA); Cy2 (Cy™2 AffiniPure donkey anti-goat IgG [H+L], 1:200, Cat. # 705-225-147, Jackson ImmunoResearch, West Grove, PA), and Cy2-streptavidin (Cy™2 AffiniPure donkey anti-biotin IgG [H+L], 1:600, Cat. # 016-220-084, Jackson ImmunoResearch, West Grove, PA). Sections were washed three times in 0.1 M PBS, dehydrated, and mounted in DPX and then cover-slipped.
Confocal microscopy, image analysis, and quantification of cell size
All images were captured on a Carl Zeiss Microscopy (Carl Zeiss, LSM 780 Confocal Microscope System, Oberkochen) with 63X/1.32 oil immersion. Optical sections were acquired at the digital size of 1024 × 1024 pixels and averaged of eight times to reduce noise. Images were improved and analyzed using the ZenZ Blue Edition Software (Carl Zeiss GmbH, Gottingen). To determine the percentage of ATF2 immunoreactivity in neurons in each rat, four to five sections of the individual L4/L5/L6 DRGs at 3, 7, 14, and 21 days post-injury were selected randomly, and 800 to 1200 neuron profiles were counted. The average percentage of positive neurons was calculated by dividing the number of ATF2+ neurons by the total number of NeuN+ neurons × 100. For the size–frequency histogram data, measurements of the area of NeuN+ and ATF2+ neurons in L4, L5, and L6 DRGs were performed using a computerized image analysis system (Image J software), and only neurons with clearly visible nucleus (NeuN+) were used for quantification. To distinguish cell size specific changes, we divided the DRG neurons into small- (<600 µm2), medium- (600–1200 µm2), and large- (>1200 µm2) sized neurons according to their cross-sectional area. Results are reported as the percentage of labeled cells within each size population according to previously established size criteria. 23 At least 300 ATF2+ and NeuN+ neuron profiles were measured by section of each experimental group. In addition, Image J software was used to calculate the ATF2 mean fluorescence intensity in L4 to L6 spinal dorsal horn from at least five slices from a group of four rats.
Semi-quantitative nested reverse transcription-polymerase chain reaction
Independent groups of four rats each were used to determine mRNA in spinal cord and DRG. Total RNA was isolated from the ipsilateral dorsal horn of rat lumbar enlargement (L1–S1) and from individual L4, L5, and L6 DRGs using Trizol Reagent, according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). The concentration and purity of the RNA were determined by measuring the absorbance at 260 and 280 nm in a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). Equal amounts (5 μg) of RNA from samples were reverse transcribed into first-strand complementary DNA (cDNA) using oligo-dT and M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA). The reaction was performed as follows: a RNA denaturation step at 36.5°C for 7 min, followed by the cDNA synthesis at 37°C for 1 h. Amplification of cDNA by polymerase chain reaction (PCR) was performed with specific primers for ATF2 (Cat. # sc-156017-PR that includes A set (forward and reverse) and B set (forward and reverse) primers, 427 bp product) and ATF3 (Cat. # sc-72029-PR that includes A set (forward and reverse) and B set (forward and reverse) primers, 1000 bp product). The first PCR reaction mixture contained 2.5 mM MgCl2, 10 mM dNTP, 1 U Taq DNA polymerase (Invitrogen, Carlsbad, CA), 1 µl of primer pair A, and 5 µg of first strand cDNA in a total volume of 50 μl. Optimal number of cycles within the linear range of amplification was selected. PCR conditions for amplification were as follows: 94°C for 5 min; 35 cycles of 94°C for 45 s, 58.5°C for 45 s, 72°C for 90 s, and 72°C for 7 min, using a Mastercycler DNA Engine Thermal Cycler (Eppendorf, Germany). The second PCR reaction mixture contained 2.5 mM MgCl2, 10 mM dNTP, 1 U Taq DNA polymerase (Invitrogen, Carlsbad, CA), 1 µl of primer pair B, and 5 µl of first PCR product in a total volume of 50 μl. To ensure that equal amounts of reverse-transcribed RNA were added to the PCR, the β-actin housekeeping gene was amplified using oligonucleotides previously reported. 24 PCR products stained with ethidium bromide were analyzed on 1% agarose gel using the Chemidoc XRS + imaging system (Bio-Rad, Hercules, CA). Bands were quantified by scanning densitometry using Image Lab 5.0 software (Bio-Rad, Hercules, CA).
Experimental design
Protein expression
In order to determine the role of spinal ATF2 or ATF3 in development and maintenance of neuropathic pain, we first assessed whether ATF2 was expressed in relevant sites for nociceptive transmission. For this, we determined ATF2 or ATF3 protein expression by western blotting in L4, L5, and L6 DRG and in the ipsilateral dorsal portion of the spinal cord of rats at 3, 7, 14, and 21 days post-injury (n = 4). It is considered that neuropathic pain develops from one to seven days after ligation. Based on this, we took samples in the period of pain development (3 and 7 days) and once it was established (14 and 21 days).
siRNA treatments
In order to determine the role of ATF2 and ATF3 in neuropathic pain, three groups of spinal nerve ligated (18 days post-injury) animals (n = 6–8 each) received a daily intrathecal injection (10 μl each) of ATF2 (1 µg/24 h, ATF2 siRNA, Cat. # sc-156017), ATF3 siRNA (1 µg/24 h, ATF3 siRNA, Cat. # sc-72029), or siRNA scramble (siRNA scramble control, Cat. # sc-37007) (Santa Cruz Biotechnology, Dallas, TX). siRNA and scrambled control doses were prepared immediately before administration with a transfection reagent (siRNA transfection reagent, Cat. # sc-29528, Santa Cruz Biotechnology, Dallas, TX), according to the manufacturer’s instructions. Injections were given daily for three consecutive days starting 18 days post-injury. Tactile allodynia and thermal hyperalgesia were determined 24 h later and then daily for two additional days until day 21. However, since the antiallodynic or antihyperalgesic effect of treatments was still high at that time, we continued assessing the effect for the next five days. By this time, most of the effects of treatments have returned to baseline. An independent group of rats (n = 4, treated with ATF3 siRNA) was sacrificed 21 days post-injury to obtain tissue samples. In these tissues, we evaluated ATF3 mRNA expression in order to assess the efficiency of Atf3 gene knockdown. Since ATF2 is constitutively expressed, efficiency of Atf2 gene knockdown was determined in naïve animals (n = 4, treated with ATF2 siRNA). mRNA analysis in each experimental condition was performed for duplicate.
Anti-ATF antibody treatments
In order to further determine the participation of ATF2 and ATF3 in the maintenance of neuropathic pain, spinal nerve ligated (18 days post-injury, n = 6–8 each) animals received three intrathecal injections (10 μl) of anti-ATF2 or anti-ATF3 antibodies (10 µg/24 h) or 1 M PBS with 0.1% triton X-100 (antibody vehicle). Baseline withdrawal thresholds to thermal or mechanical stimuli were evaluated immediately before intrathecal administration of antibodies. Injections were given daily for 3 consecutive days starting 18 days post-injury. Tactile allodynia and thermal hyperalgesia were determined 24 h later and then daily for two additional days until day 21. However, since the antiallodynic or antihyperalgesic effect of treatments was still high at that time, we continued assessing the effect for an additional one day. By this time, all effects of treatments had returned to baseline.
Localization of ATF2 in the DRG and spinal cord
Based on our molecular and behavioral results, we sought to investigate the distribution of ATF2 at the DRGs and dorsal horn spinal cord. For this purpose, we used six groups of four rats each: naïve, sham-operated, and spinal nerve ligated rats at 3, 7, 14, and 21 days post-injury. Tissue samples were obtained as previously described (see Immunohistochemistry section) and processed for immunofluorescence analysis.
Data analysis and statistics
Behavioral data are expressed as mean (n = 6–8) ± SEM of the hind paw withdrawal threshold or withdrawal latency. Curves were constructed by plotting the paw withdrawal threshold or withdrawal latency as a function of time. Protein and mRNA expression data are expressed as ATF2 or ATF3 relative expression normalized against β-actin. Data are the mean ± SEM of four independent animals. Groups of four rats each were used for the immunohistochemical experiments.
Statistical differences between groups were determined by one- or two-way analysis of variance, followed by the Tukey test. P values less than 0.05 were considered significant.
Results
Spinal nerve injury increases ATF2 and ATF3 expression in DRGs and spinal cord
Although ATF2 is widely expressed in many tissues, its role in neuropathic pain is uncertain. Therefore, we first assessed whether ATF2 was expressed in relevant sites for nociceptive transmission. ATF2 was present in naïve and sham-operated animals (n=4). Moreover, spinal nerve ligation enhanced ATF2 expression in injured DRGs (L5 and L6) and spinal cord at 14 and 21 days post-injury but not in uninjured DRG (L4) nor at 3 and 7 days post-injury (n=4, Figure 1a to d).
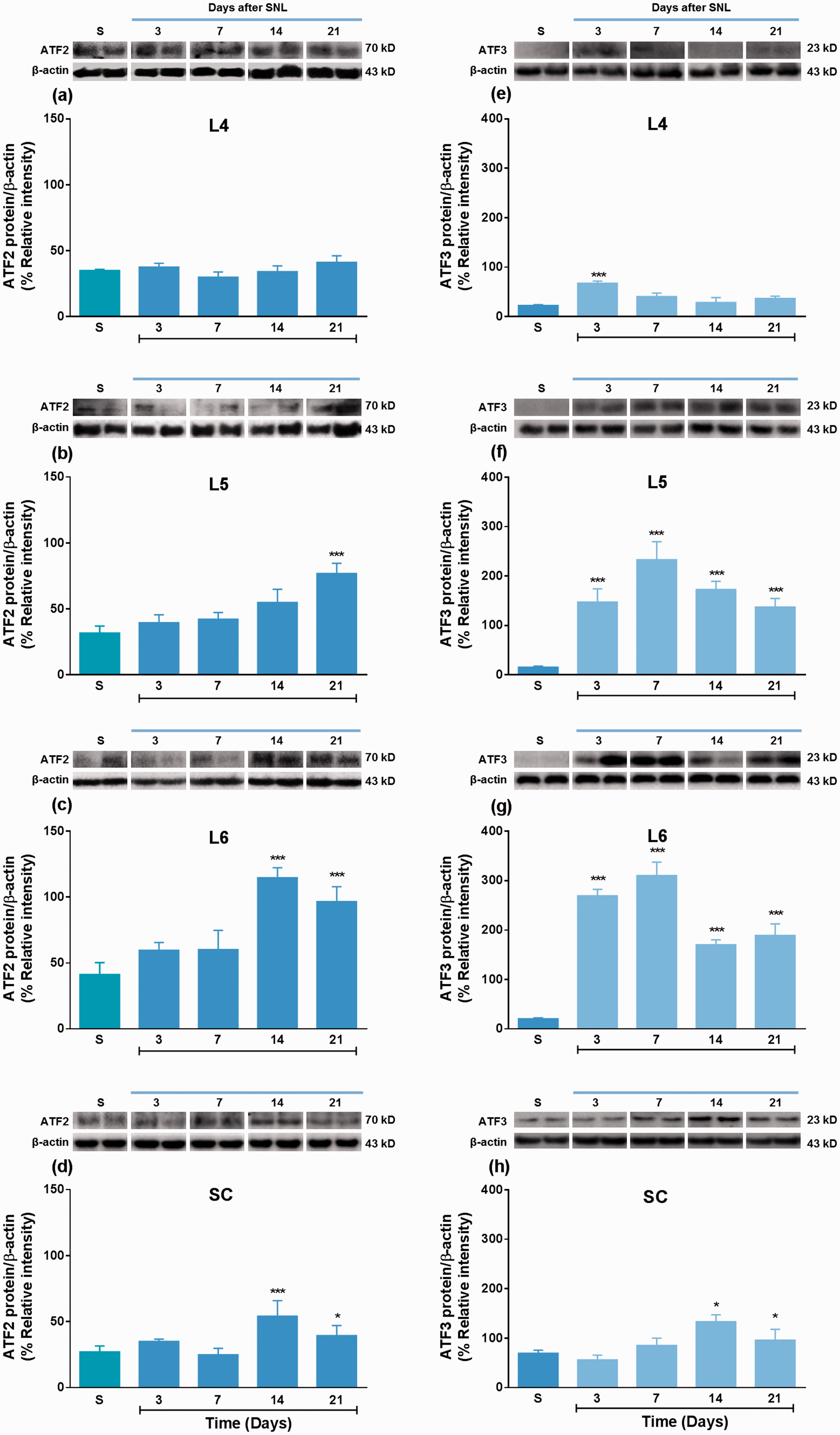
Nerve injury enhances ATF2 and ATF3 expression at the dorsal root ganglia (DRG) and dorsal spinal cord. Western blot analysis of ATF2 (panels a to d) and ATF3 (panels e to h) in uninjured (L4) and injured (L5 and L6) DRGs and dorsal horn spinal cord obtained from sham-operated and spinal nerve ligated (SNL) rats. Data were normalized against β-actin and are expressed as the mean ± SEM of four independent rats. Insets in (a) to (d) and (e) to (h) show representative blots obtained with ATF2, ATF3, and β-actin primary antibodies, which revealed bands around 70, 23, and 43 kDa, respectively. *P < 0.05 and ***P < 0.001, significantly different from the sham (S) group, as determined by one-way ANOVA followed by the Tukey test.
ATF3 protein participates in regeneration processes and is widely expressed in injured neurons.25,26 However, its involvement in pain is unknown. ATF3 expression in L4, L5, and L6 DRGs of sham animals was low (Figure 1e to g). In contrast, ATF3 was found in the dorsal portion of the spinal cord (Figure 1h). As expected, we found that spinal nerve injury increased ATF3 expression in injured DRGs (L5 and L6) from 3 to 21 days post-injury (Figure 1f and g). ATF3 expression remained elevated until day 21 post-injury. In contrast, we found a transient increase of ATF3 expression at day 3 post-injury in uninjured DRG (L4) (Figure 1e). Interestingly, we found that nerve injury enhanced ATF3 expression at the spinal cord 14 to 21 days post-injury (Figure 1h).
Localization of ATF2 in the DRG and spinal cord
We found that ATF2 was constitutively expressed in the nucleus of L4, L5, and L6 DRG neurons in naïve and sham-operated rats (Table 1). Moreover, nerve injury enhanced ATF2 immunoreactivity in the cell nucleus of injured L5 and L6 DRGs at 7 days after injury, and in the nucleus and cytoplasm at 14 and 21 days post-surgery (Table 1, Figure 2f to o), but not in uninjured L4 DRG (Figure 2a to e). In contrast, sham surgery, compared to naïve animals, did not modify ATF2 immunoreactivity (Table 1). Interestingly, nerve injury reduced ATF2 immunoreactivity in cell nucleus and increased that around neuronal bodies three days after surgery, which suggests its presence in satellite glial cells (Figure 2l). Moreover, we observed ATF2 immunoreactivity in the spinal cord of naïve and sham-operated rats. Of note, nerve injury increased ATF2 immunoreactivity in the superficial and deep laminae of the spinal cord at 7, 14, and 21 days post-surgery (Figure 2p to t).
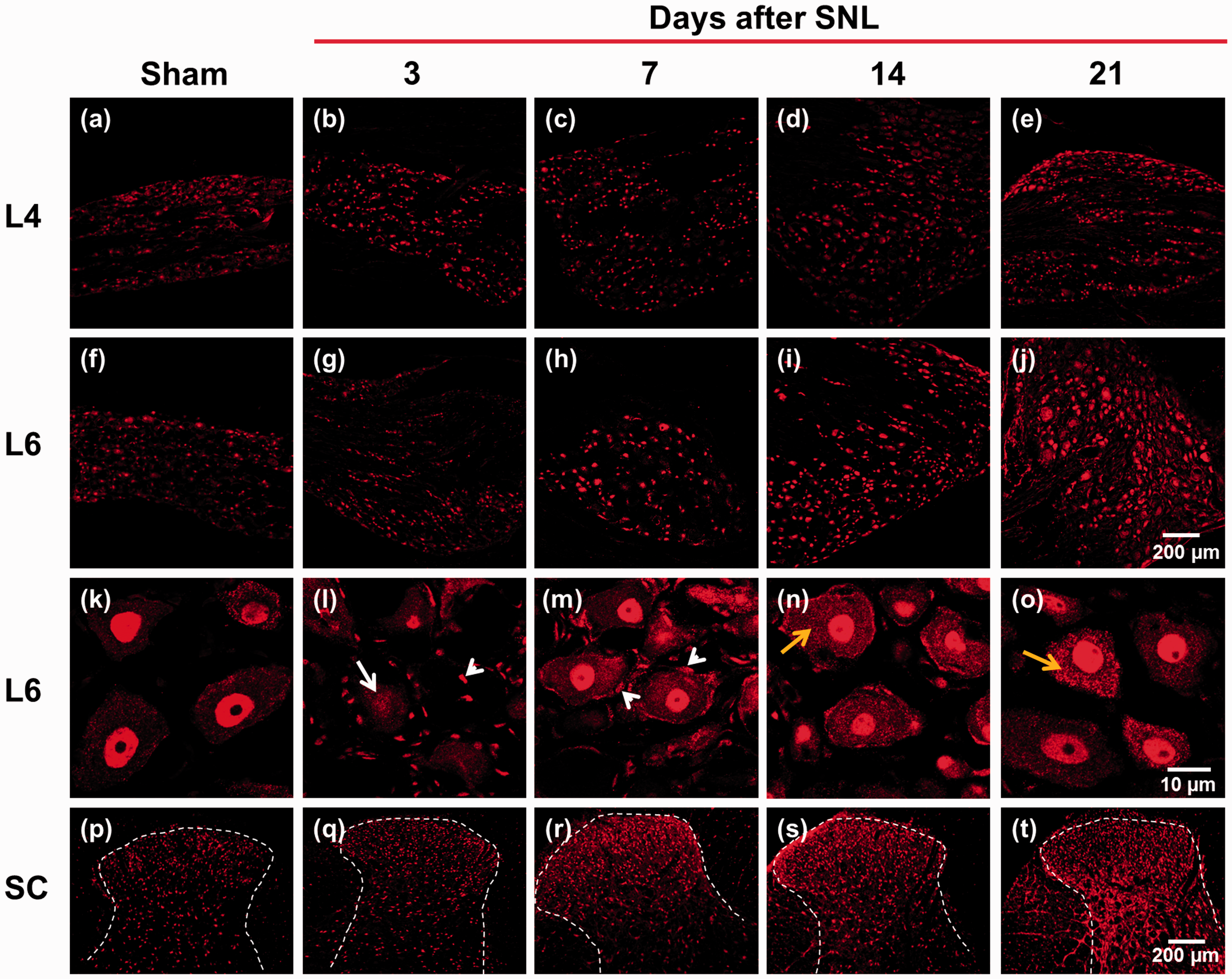
Spinal nerve ligation (SNL) enhances ATF2 immunoreactivity at the DRG and spinal cord (SC). Representative staining of ATF2 in uninjured (L4, panels a to e) and injured (L6, panels f to o) DRGs and spinal cord (panels p to t) obtained from sham (left panels) and nerve injured rats at 3, 7, 14, and 21 days post-injury. White arrows (panel l) indicate a reduction in ATF2 immunoreactivity three days post-injury. White arrow heads (panels l and m) indicate ATF2 immunoreactivity in satellite glial cells three and seven days post-injury. Yellow arrows (panels n and o) indicate ATF2 immunoreactivity in cytoplasm 14 and 21 days post-injury. Scale bar: 200 and 10 µm.
Nerve injury increases ATF2 immunoreactivity in injured L5 and L6 DRGs but not in uninjured L4 DRG (A), and nerve injury enhances ATF2 fluorescence intensity at the spinal cord (B).

Data in A are expressed as % of neurons positive to ATF2/neurons positive to NeuN. Data in B are expressed as the mean fluorescence intensity of ATF2. *P < 0.05, **P < 0.01, and ***P < 0.001 significantly different versus the naive group, by one-way ANOVA followed by the Tukey test. ATF: activating transcription factors.
Effect of the ATF2 siRNA and anti-ATF2 antibody on nerve injury-induced tactile allodynia and thermal hyperalgesia
Once we established that ATF2 is expressed in DRG and spinal cord, and that nerve injury up-regulates this protein, we sought to investigate whether ATF2 is involved in the maintenance of neuropathic pain. For this, we used a siRNA directed against ATF2 and an anti-ATF2 antibody. Repeated intrathecal administration of the ATF2 siRNA (three times, 1 μg/24 h), but not scramble control siRNA (three times, 1 μg/24 h), decreased the Atf2 gene expression (ATF2 mRNA) in L4, L5, and L6 DRGs and dorsal spinal cord of naïve rats (Supplementary Figure 1). Moreover, ATF2 knockdown by ATF2 siRNA or anti-ATF2 antibody produced transient mechanical hypersensitivity but they did not modify withdrawal latency to thermal stimuli in naïve rats (Supplementary Figure 2). Repeated intrathecal administration with the ATF2 siRNA (three times, 1 μg/24 h) or anti-ATF2 antibody (three times, 10 μg/24 h, starting at day 18 after nerve injury) partially and transiently reversed tactile allodynia (Figure 3a). Treatments reached the maximal effects 24 to 48 h after the last injection, and these effects disappeared three days after the last injection. In addition, both treatments reversed thermal hyperalgesia in nerve-injured rats (Figure 3b). Of note, the anti-hyperalgesic effect of ATF2 siRNA or anti-ATF2 antibody reached a maximal effect and lasted longer (five days after the last injection) than those observed in the antiallodynic effect (Figure 3b). Both, the scramble control siRNA and the antibody vehicle (PBS) did not affect baseline threshold to mechanical and thermal stimuli (Figure 3).
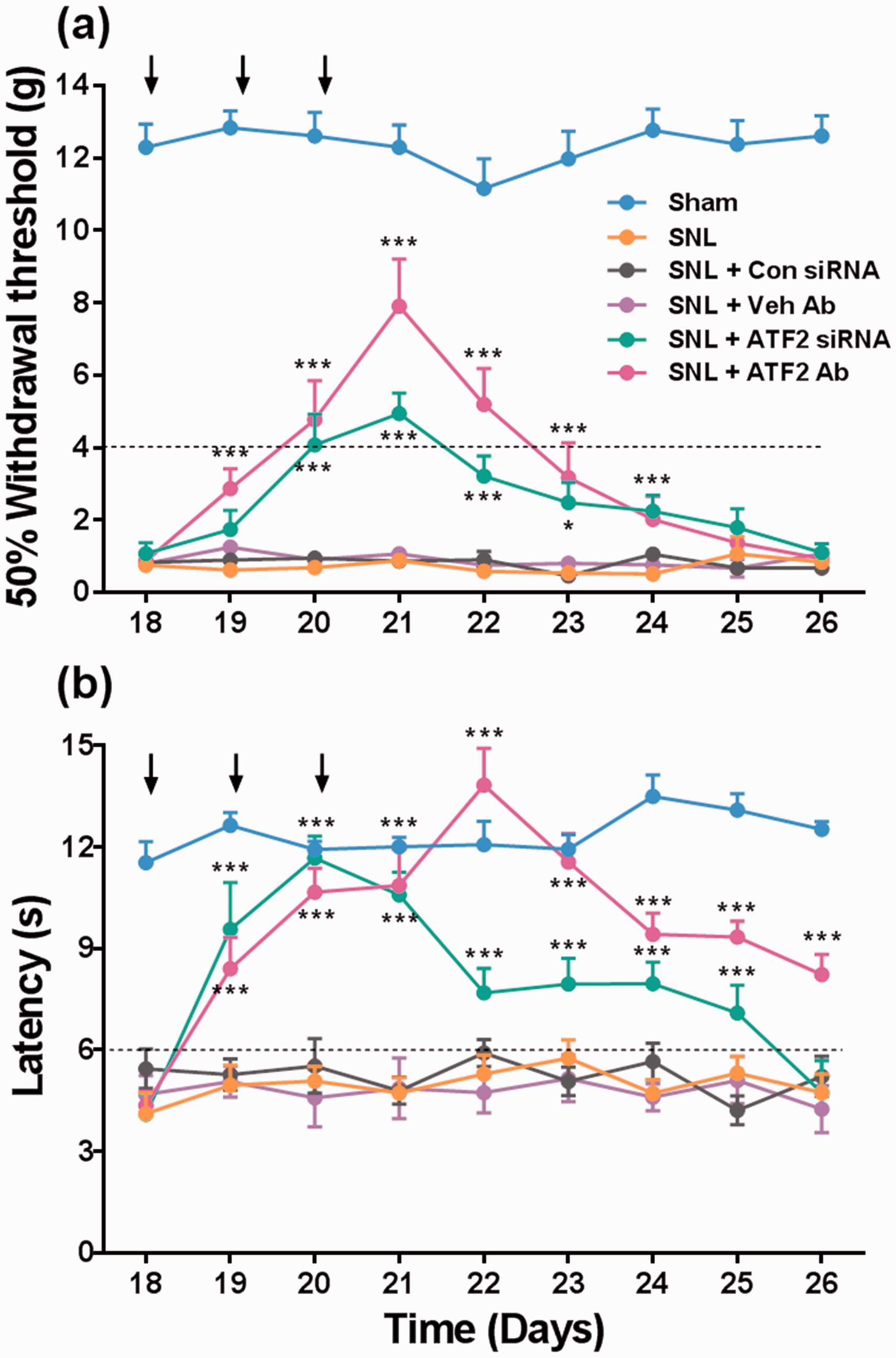
Spinal ATF2 is involved in the maintenance of spinal nerve ligation (SNL)-induced neuropathic pain. Effect of repeated intrathecal injection of the ATF2 siRNA (1 μg) or anti-ATF2 antibody (ATF2 Ab, 10 μg) on tactile allodynia (panel a) and thermal hyperalgesia (panel b) in rats previously (18 days) subjected to SNL. Arrows in panels a and b indicate daily injection of the ATF2 siRNA, anti-ATF2 antibody, control siRNA (Con siRNA), or antibody vehicle (Veh Ab). Data are expressed as the mean (n = 6–8) ± SEM. *P < 0.05 and ***P < 0.001 significantly different from the SNL group, by two-way ANOVA followed by the Tukey test.
Effect of the ATF3 siRNA and anti-ATF3 antibody on nerve injury-induced tactile allodynia and thermal hyperalgesia
As previously reported,11,14 nerve injury up-regulated ATF3 expression in DRG and spinal cord. However, whether these changes are involved in the maintenance of neuropathic pain is unknown. In order to evaluate this, we used the ATF3 siRNA or anti-ATF3 antibody. Repeated intrathecal administration of the ATF3 siRNA (three times, 1 μg/24 h) decreased nerve injury-induced up-regulation of ATF3 mRNA in injured DRGs (L5 and L6) and dorsal spinal cord (Supplementary Figure 3). In contrast, scramble control siRNA (three times, 1 μg/24 h) did not modify ATF3 mRNA expression in DRG or spinal cord (data not shown). ATF3 knockdown did not modify the withdrawal thresholds to mechanical and thermal stimuli in naïve rats (Supplementary Figure 4). Unlike ATF2, ATF3 knockdown induced by ATF3 siRNA slightly diminished tactile allodynia but not thermal hyperalgesia in neuropathic rats. In contrast, the anti-ATF3 antibody (three times, 10 μg/24 h) slightly diminished thermal hyperalgesia but not tactile allodynia (Figure 4).
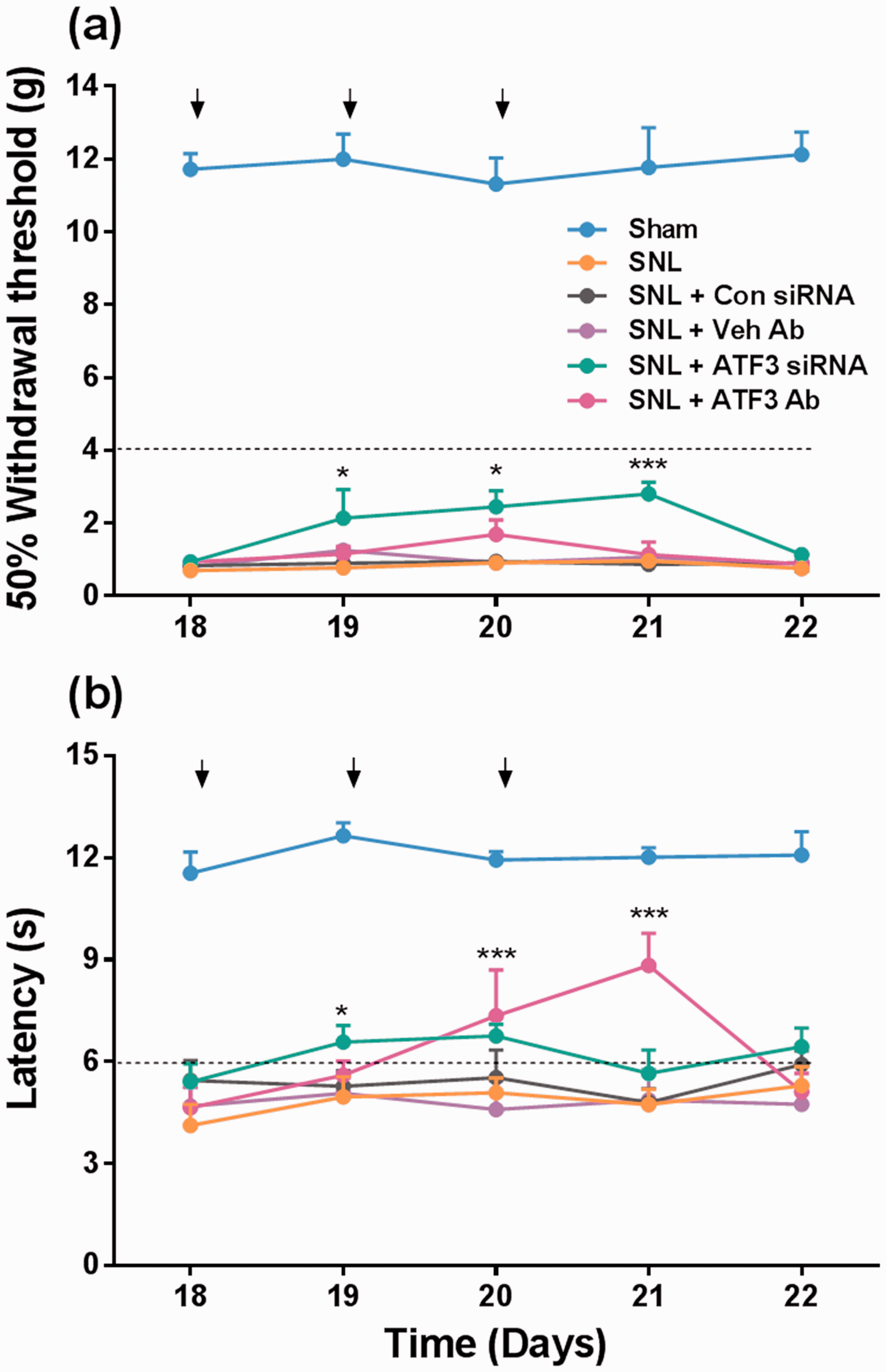
Spinal ATF3 does not participate in the maintenance of spinal nerve ligation (SNL)-induced neuropathic pain. Effect of repeated intrathecal injection of the ATF3 siRNA (1 μg) or anti-ATF3 antibody (ATF3 Ab, 10 μg) on tactile allodynia (panel a) and thermal hyperalgesia (panel b) in rats previously (18 days) subjected to SNL. Arrows in panels a and b indicate daily injection of the ATF3 siRNA, anti-ATF3 antibody, control siRNA (Con siRNA), or antibody vehicle (Veh Ab). Data are expressed as the mean (n = 6–8) ± SEM. *P < 0.05 and ***P < 0.001 significantly different from the SNL group, by two-way ANOVA followed by the Tukey test.
Localization of ATF2 in DRG and spinal cord
In order to further investigate the cellular localization of ATF2, we performed multiple immunolabeling experiments in defined cellular compartments of the DRG. These included neuronal bodies (NeuN), peptidergic (CGRP), and non-peptidergic (IB4) neurons, as well as satellite glial cells (GFAP). The specificity of the primary antibodies was confirmed through the immunostaining in the absence of the respective primary antibodies. Moreover, the tissue integrity was evaluated by using the Nissl staining (data not shown). As expected, ATF2 immunoreactivity co-localized with NeuN immunoreactivity in L4, L5, and L6 DRGs of naïve rats (Figure 5a, e, and i). Furthermore, ATF2 immunoreactivity was found mainly in non-peptidergic (IB4+) (Figure 5c, g and k) but also in peptidergic (CGRP+) neurons (Figure 5b, f, and j). ATF2 was constitutively expressed in small-, medium-, and large-size neurons in L4, L5, and L6 DRGs of naïve rats (Figure 6a to c). However, ATF2 was found mainly in small and medium DRG neurons. ATF2 immunoreactivity was not found in satellite glial cells (GFAP+).
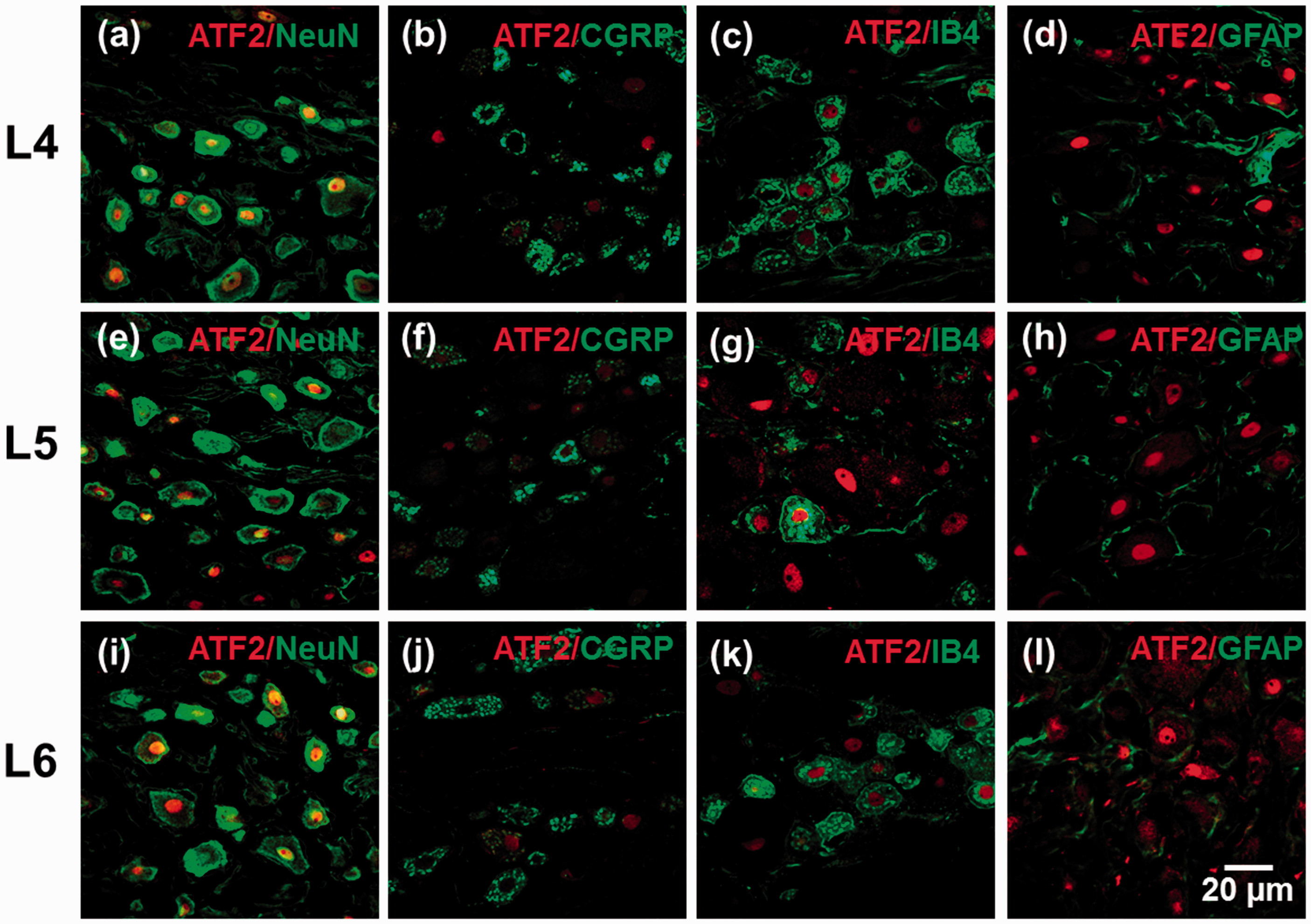
ATF2 is expressed in L4, L5, and L6 DRG neurons. Distribution of ATF2 in L4 (panels a to d), L5 (panels e to h), and L6 (panels i to l) DRGs of naïve rats. All panels: ATF2 (red), NeuN (green), CGRP (green), IB4 (green), GFAP (green), ATF2, and NeuN co-expression (yellow) in small-, medium-, and large-sized neurons. Scale bar: 20 µm.

Size-frequency distribution of ATF2+ and NeuN+ neurons in L4 (panel a), L5 (panel b), and L6 (panel c) DRGs in naïve rats. Data were obtained from four to five sections of four animals using ImageJ software to manually measure cross-sectional area of neurons in 14-µm-thick sections. Data are plotted as total number of neurons.
In order to investigate the cellular localization of ATF2 in the spinal cord, we performed multiple immunolabeling experiments in spinal cord. These included neuronal bodies (NeuN), peptidergic (CGRP), non-peptidergic (IB4), microglia (OX-42), and astrocytes (GFAP). ATF2 immunoreactivity was found mainly at the superficial laminae (I–III) of the spinal cord of naïve rats (Figure 7). In this condition, ATF2 immunoreactivity was found in dorsal horn neurons (NeuN+) (Figure 7a and b). Unlike DRG, we were not able to find ATF2 immunoreactivity in non-peptidergic nor peptidergic neurons (Figure 7c and d). ATF2 immunoreactivity was not found in astrocytes or microglia (Figure 7e and f).

ATF2 is expressed at the dorsal horn spinal cord. Distribution of ATF2 at the dorsal horn spinal cord of naïve rats. All panels: ATF2 (red, panel b), NeuN (green, panel b), CGRP (green, panel c), IB4 (green, panel d), GFAP (green, panel e), OX-42 (green, panel f), ATF2, and NeuN co-expression (yellow). Scale bar: 50 µm.
Discussion
ATF2 and neuropathic pain
Here we demonstrated that ATF2 participates in nerve injury-induced tactile allodynia and thermal hyperalgesia in rats. First, we found that spinal nerve ligation increased ATF2 expression in DRG neurons and spinal cord of rats. Since nerve injury mainly enhanced ATF2 expression at 14 and 21 days, it is likely that this transcription factor may participate in the maintenance of neuropathic pain. In this study, we only measured ATF2 but not p-ATF2. It is known that ATF2 is a downstream substrate for the P38-MAPK. 27 As P38-MAPK contributes to hypersensitivity in neuropathic pain models, 28 it is possible that nerve injury could lead to the increase of ATF2 and p-ATF2. Our data agree with previous studies showing that sciatic nerve axotomy and complete Freund’s adjuvant increase expression of p-ATF2 at the DRG and spinal cord, respectively.9,10 In contrast, there is evidence that transection of the optic, vagal, and facial nerves rapidly decreased ATF-2 in axotomized neurons while that ultraviolet irradiation reduced ATF2 expression in cultured adult DRG neurons. 29 Differences could be due to site of injury or preparation of DRG neurons. Second, we demonstrated that intrathecal injection of the ATF2 siRNA or anti-ATF2 antibody transiently reversed nerve injury-induced tactile allodynia and thermal hyperalgesia. This result further supports our suggestion that ATF2 plays a pronociceptive role in the maintenance of neuropathic pain. To the best of our knowledge, this is the first report about the function of ATF2 in the maintenance of neuropathic pain in rats. The antinociceptive effects of these tools could be due to the knockdown of the Atf2 gene, as ATF2 siRNA reduced ATF2 mRNA expression in naïve animals. Of note, ATF2 siRNA or anti-ATF2 antibody induced tactile allodynia but not thermal hyperalgesia in naive animals. Although the mechanism of this effect is unknown, data suggest that ATF2 could have a tonic antinociceptive role in normal conditions, whereas that this transcription factor has indeed a pronociceptive role in pathological conditions (nerve injury). This is not uncommon in pain, as it is the case of GABAA receptors and KCC2.30,31 The putative protective effect of ATF2 in naïve animals agrees with a previous study showing that disruption of the murine ATF2 locus leads to neonatal neuronal cell death. 8
In contrast to ATF2, ATF3 is an inducible transcription factor expressed at very low levels in intact neuronal tissue.11,17 However, there is evidence that nerve injury increases ATF3 expression.11,14,17,32 In fact, this transcription factor is currently considered as a nerve injury marker.14,33 Actually, it has been speculated that ATF3 could contribute to formalin-induced long-lasting behavioral effects. 11 As expected, we found that nerve injury up-regulated ATF3 expression mainly in L5 and L6 DRGs and spinal cord. Interestingly, nerve injury induced a small and transient increase of ATF3 expression in non-injured L4 DRG (three days post-injury). These results partially agree with a previous observation showing that L5 spinal nerve ligation slightly induces ATF3 expression in L4 DRG from day 1 to 28 after injury. 34 Differences could be due to the different techniques to determine ATF3 expression (western blot versus in situ hybridization histochemistry). Unlike ATF2, repeated intrathecal administration of the ATF3 siRNA did not modify nerve injury-induced tactile allodynia nor thermal hyperalgesia while the anti-ATF3 antibody slightly modified thermal hyperalgesia but not tactile allodynia. Lack of effect of the ATF3 siRNA was not due to a failure to silence the Atf3 gene, as this siRNA was able to diminish nerve injury-induced ATF3 mRNA up-regulation. Therefore, our results suggest that although ATF3 is up-regulated, it seems to play a minor role in spinal nerve ligation-induced tactile allodynia. However, based on our results, ATF3 could have a role in thermal hyperalgesia. This result agrees with a previous suggestion about the participation of ATF3 in formalin-induced long-lasting hypersensitivity. 11 The use of other models of neuropathic pain could help to shed light on this point.
Interestingly, we found that ATF3 is not only expressed in damaged neuronal tissue but also in the spinal cord neurons. There is evidence that damage to the peripheral nerves leads to the loss of projection neurons in the superficial dorsal horn probably due to glutamate-mediated excitotoxicity, 35 which could be one of the causes that lead to ATF3 up-regulation at the spinal cord. This hypothesis will need substantial experiments. ATF3 participates in peripheral nerve regeneration processes as it favors the expression of c-Jun, HSP27, SPRR1A, and damage-induced neuronal endopeptidase,25,26,36,37 which in turn reduce neuronal death and favor neurite growth.15,16,38 Notwithstanding these effects, ATF3 seems to play a minor role in the behavioral effects of neuropathic pain.
Localization of ATF2 in DRG and spinal cord
ATF2 immunoreactivity was found in the cell nucleus of L4, L5, and L6 DRG neurons as well as in the spinal cord of naïve and sham-operated rats indicating that this transcription factor is constitutively expressed in sensory neurons. Nerve injury reduced ATF2 immunoreactivity in the nucleus while it enhanced that in satellite glial cells at three days post-injury. Mounting evidence suggests that activation of satellite glial cells indeed participate in neuropathic pain.39–41 Thus, the transient presence of ATF2 in these cells may have a role in neuropathic pain. Our data partially agree with other observations indicating that stimulation of adult DRG neurons by doxorubicin or ultraviolet irradiation (in vitro) and axotomy (in vivo) induce a rapid (2–24 h) and consistent (3–20 days) down-regulation of ATF2.29,42,43 In contrast, we found that nerve injury enhanced ATF2 immunoreactivity in L5 and L6, but not L4, DRG and spinal cord in a time-dependent manner (7–21 days). Differences could be due to the sites of nerve injury. Interestingly, we found that nerve injury enhanced ATF2 immunoreactivity into the cytoplasm of L6 DRG neurons. To our knowledge, this is the first report about the translocation of ATF2 from cellular nucleus to cytoplasm by nerve injury. The function of ATF2 in cytoplasm is unknown. Several studies have reported that nerve injury increases c-Jun expression at the DRG and spinal cord.44–46 Moreover, ATF2 binds to c-Jun in order to modulate expression of several targets.47,48 For example, there is evidence that c-Jun modulate the expression of vasoactive intestinal peptide and neuropeptide Y in cultured DRG neurons in neuropathic rats. 49 Thus, the heterodimer ATF2/c-Jun could promote nerve injury-induced nociception through the enhanced expression of vasoactive intestinal peptide and neuropeptide Y in DRG and spinal cord. However, ATF2 could have other targets in order to promote nociception in neuropathic rats. In this sense, ATF2 may bind to the promoter site of several genes involved in neuropathic pain (Asic2, Cacna2d1, Clcn1/2/3/6/7, Best1/4, and Trpv1/2/3/4). 50
ATF2 immunoreactivity was found in the cell nucleus of L4-L6 DRG neurons and spinal cord of sham/naïve rats suggesting that this transcription factor is constitutively expressed in DRG and spinal cord neurons. Interestingly, ATF2 was mainly found in non-peptidergic neurons. In addition, ATF2 was found mainly in small and medium DRG neurons. This distribution of ATF2 in nociceptive fibers would suggest that this transcription factor is involved in pain processing. Accordingly, our molecular and behavioral results suggest that ATF2 has a pronociceptive role in neuropathic pain. This seems to be the first report about the distribution of ATF2 in DRG neurons. Our immunofluorescence results along with the western blotting studies support a role of ATF2 in the spinal nociceptive processing.
Conclusion
Our results suggest that ATF2, but not ATF3, localized in DRG and dorsal horn spinal cord plays a pronociceptive role in pathological conditions (nerve injury). In contrast, ATF2 seems to play an antinociceptive role in normal conditions. Our data indicate that spinal ATF2 participates in the maintenance of nerve injury-induced neuropathic pain.
Footnotes
Author Contributions
Ana Belén Salinas-Abarca, Isabel Velazquez-Lagunas, and Úrzula Franco-Enzástiga participated in the design, acquisition of data or analysis, and interpretation of data. All of them performed experiments. Ana Belén Salinas-Abarca drafted the manuscript. Jorge Elias Torres-Lopez and Héctor Isaac Rocha-González participated in the design, acquisition of data or analysis, and interpretation of data and corrected the first draft. Vinicio Granados-Soto conceived the idea, designed the experiments, got extramural funds, and corrected the final draft. All authors approved the version to be published.
Acknowledgments
This work is part of the PhD dissertation of Ana Belen Salinas-Abarca. The authors are grateful with Dr. Claudia González-Espinosa (Cinvestav, Unidad Coapa) for the generous gift of the PageRuler Plus Prestained Protein Ladder and also greatly appreciate the technical assistance of Guadalupe C. Vidal-Cantú and José Rodolfo Fernández.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Isabel Velazquez-Lagunas (fellowship 424047), Úrzula Franco-Enzástiga (fellowship 338449), and Ana Belen Salinas-Abarca (fellowship 277977) are Conacyt fellows. This study was partially supported by Conacyt (grant CB-2012/179294 to VG-S) and UJAT (grant OP/PFCE-2018-27MSU0018V-07-01 to JET-L).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.