
Abstract
Chemokines-mediated neuroinflammation in the spinal cord plays a critical role in the pathogenesis of neuropathic pain. Chemokine CXCL9, CXCL10, and CXCL11 have been identified as a same subfamily chemokine which bind to CXC chemokine receptor 3 to exert functions. Our recent work found that CXCL10 is upregulated in spinal astrocytes after spinal nerve ligation (SNL) and acts on chemokine receptor CXCR3 on neurons to contribute to central sensitization and neuropathic pain, but less is known about CXCL9 and CXCL11 in the maintenance of neuropathic pain. Here, we report that CXCL9 and CXCL11, same as CXCL10, were increased in spinal astrocytes after SNL. Surprisingly, inhibition of CXCL9 or CXCL11 by spinal injection of shRNA lentivirus did not attenuate SNL-induced neuropathic pain. In addition, intrathecal injection of CXCL9 and CXCL11 did not produce hyperalgesia or allodynia behaviors, and neither of them induced ERK activation, a marker of central sensitization. Whole-cell patch clamp recording on spinal neurons showed that CXCL9 and CXCL11 enhanced both excitatory synaptic transmission and inhibitory synaptic transmission, whereas CXCL10 only produced an increase in excitatory synaptic transmission. These results suggest that, although the expression of CXCL9 and CXCL11 are increased after SNL, they may not contribute to the maintenance of neuropathic pain.
Introduction
Chemokines are a family of closely related chemoattractant cytokines which promote the recruitment and activation of various immune cells. 1 Based on the conserved cysteine motifs, chemokines are classified as cysteine (C), cysteine cysteine (CC), cysteine X cysteine (CXC), and cysteine X3 cysteines (CX3C) subsets. CXCL9 (also called monokine induced by gamma interferon, Mig), CXCL10 (also called interferon gamma-induced protein 10, IP-10), and CXCL11 (also called interferon-inducible T-cell alpha chemoattractant, I-TAC) have been identified as a same subfamily chemokine which bind to a G protein-coupled receptor, CXC chemokine receptor 3 (CXCR3). 2 In the peripheral tissues, these chemokines are secreted by lymphocytes, endothelial cells, and hematopoietic progenitor cells. 3 The receptor CXCR3 is also expressed in various cells, including natural killer cells, CD4+ and CD8+ T cells, monocytes, dendritic cells, and endothelial cells.4–6 CXCR3 and its ligands have been demonstrated to play a pivotal role in the pathology of infections and autoimmune diseases.7,8
Neuroinflammation resulting from the release of inflammatory mediators from glial cells and neurons in the central nervous system has been identified to be important in the pathogenesis of neuropathic pain. 9 Several chemokines including CCL2, CXCL1, CXCL13, and CX3CL1 act on their respective receptors (CCR2, CXCR2, CXCR5, and CX3CR1) to mediate neuroinflammation in the spinal cord via different forms of neuron–glia interaction.10–13 For example, CX3CL1 and CXCL13 are expressed in spinal neurons and induce the activation of microglia and astrocytes via acting on the receptor CX3CR1 and CXCR5, respectively.10,12 In addition, chemokines CCL2 and CXCL1 are expressed in spinal astrocytes and act on CCR2 and CXCR2 in spinal neurons to increase excitatory synaptic transmission.11,13,14 The gain of excitatory synaptic transmission and loss of inhibitory synaptic transmission in dorsal horn neurons are critical mechanisms for pain sensitization.15,16 CXCL10, which is a major ligand for CXCR3 and has a dominant role in most immune responses, 17 was recently found to be highly upregulated in spinal astrocytes after spinal nerve ligation (SNL) and enhanced excitatory synaptic transmission via neuronal CXCR3 to contribute to neuropathic pain. 17 The other two ligands of CXCR3, CXCL9 and CXCL11, which have distinct kinetics and tissue expression patterns during immunoinflammatory responses,18–20 are less studied in neuropathic pain.
In the present study, we investigated whether CXCL9 and CXCL11 contribute to neuropathic pain using the well-established SNL model. Similar to the increased expression of CXCL10, 21 the mRNA and protein expression for CXCL9 and CXCL11 were also markedly upregulated in the spinal dorsal horn after SNL. However, inhibition of spinal CXCL9 or CXCL11 did not affect SNL-induced pain hypersensitivity. Intrathecal injection of CXCL9 and CXCL11 in healthy mice did not induce the hyperalgesia behaviors either. Electrophysiological recording showed that CXCL9 and CXCL11 have different effect from CXCL10 on inhibitory synaptic transmission on lamina II neurons in the spinal dorsal horn.
Materials and methods
Animals and surgery
Adult ICR (male, 6–8 weeks) mice were purchased from the Experimental Animal Center of Nantong University. All animal procedures performed in this study were reviewed and approved by the Animal Care and Use Committee of Nantong University and performed in accordance with the guidelines of the International Association for the Study of Pain. SNL was performed as previously described. 13 For sham operations, the L5 spinal nerve was exposed but not ligated.
Drugs and administration
Recombinant murine CXCL9, CXCL10, and CXCL11 were purchased from PeproTech. Intrathecal injection was made with a 30-G needle between the L5 and L6 intervertebral spaces to deliver the reagents of the CSF.
Real-time quantitative polymerase chain reaction
The total RNA of the spinal cord was extracted using Trizol reagent (Invitrogen). One microgram of total RNA was reverse-transcribed using an oligo primer according to the manufacturer’s protocol (Takara). Quantitative polymerase chain reaction (qPCR) analysis was performed in a real-time detection system (Rotor-Gene 6000, Qiagen) by SYBR green I dye detection (Takara). The detailed primer sequences for each gene (Cxcl9, Cxcl10, Cxcl11, and Gapdh) are listed in Table 1. The PCR amplifications were performed at 95°C for 30 s, followed by 40 cycles of thermal cycling at 95°C for 5 s and 60°C for 45 s. Gapdh was used as an endogenous control to normalize differences. Melt curves were performed on completion of the cycles to ensure that nonspecific products were absent. Quantification was performed by normalizing Ct (cycle threshold) values with Gapdh Ct and analyzed with the 2−△△CT method.
Primer sets used in qPCR.

ELISA
Mouse CXCL9 ELISA kit was purchased from R&D Systems. Animals were transcardially perfused with phosphate buffered saline (PBS). Spinal cord tissues were homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). For each reaction in a 96-well plate, 100 μg of proteins were used, and ELISA was performed according to the manufacturer’s protocol. The standard curve was included in each experiment.
Western blot
Protein samples were prepared in the same way as for ELISA analysis. Protein samples (30 μg) were separated on SDS–PAGE gel and transferred to nitrocellulose blots. The blots were blocked with 5% milk and incubated overnight at 4°C with antibody against CXCL11 (1:500, BioRad, China) and pERK (phosphorylated extracellular signal-regulated kinase; 1:500; Cell Signaling Technology). For loading control, the blots were incubated with GAPDH antibody (mouse, 1:20,000, Millipore). These blots were further incubated with horseradish peroxidase-conjugated secondary antibody, developed in ECL solution, and exposed onto Hyperfilm (Millipore). Specific bands were evaluated by apparent molecular size. The intensity of the selected bands was analyzed using ImageJ software (National Institutes of Health).
Immunohistochemistry
Animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS followed by 4% paraformaldehyde. After the perfusion, the L5 spinal cord segment was removed and post fixed in the same fixative overnight. Spinal cord sections (30 μm, free-floating) were cut in a cryostat and processed for immunofluorescence as previously described. 22 The sections were first blocked with 5% donkey serum for 2 h at room temperature, then incubated overnight at 4°C with the following primary antibodies: CXCL9 (Rabbit, 1:500, Bio-Rad), CXCL11 (Rabbit, 1:500, Bio-Rad), glial fibrillary acidic protein (GFAP; mouse, 1:5000, Millipore), NeuN (mouse, 1:1000, Millipore), and CD11b (mouse, 1:100, Serotec). The sections were then incubated for 1 h at room temperature with Cy3-conjugated or fluorescein isothiocyanate-conjugated secondary antibodies (1:400, Jackson ImmunoResearch). For double immunofluorescence, sections were incubated with a mixture of different primary antibodies followed by a mixture of fluorescein isothiocyanate-conjugated and Cy3-conjugated secondary antibodies. The specificity of CXCL9 and CXCL11 primary antibody was tested by preabsorption experiment. In brief, spinal cord sections were incubated with a mixture of CXCL9 or CXCL11 primary antibody and the corresponding blocking peptide (10 μg/ml; Bio-Rad) overnight, followed by secondary antibody incubation. The stained sections were examined with a Leica fluorescence microscope, and images were captured with a CCD Spot camera.
Lentiviral vectors production and intraspinal injection
The shRNAs targeting the sequence of mice CXCL9 (Gene Bank Accession: NM_008599.4), CXCL10 (NM_021274.2), or CXCL11 (NM_019494.1) were designed. An additional scrambled sequence was also designed as a negative control (NC). The recombinant lentivirus containing Cxcl9 shRNA (LV-Cxcl9 shRNA, 5′-TCG AGG AAC CCT AGT GAT A-3′), Cxcl10 shRNA (LV-Cxcl10 shRNA, 5′-GCT GCA ACT GCA TCC ATA T-3′), Cxcl11 shRNA (LV-Cxcl11 shRNA, 5′-TCT GTA ATT TAC CCG AGT A-3′), or NC shRNA (LV-NC, 5′-TTC TCC GAA CGT GTC ACG T-3′) was packaged using pGCSIL-GFP vector by Shanghai GeneChem. To test the knockdown effect, the Cxcl9-, Cxcl10-, or Cxcl11-expressing plasmid and the corresponding shRNA plasmid were transfected to HEK293 cells. Two days after transfection, the cells were harvested and subjected to quantitative RT-PCR. The intraspinal injection was performed as described previously. 13 In brief, animals were anesthetized and underwent hemilaminectomy at the L1-L2 vertebral segments. After exposure of the ipsilateral spinal cord, each mouse received two injections (0.4 μL, 0.8 mm apart and 0.5 mm deep) of the lentivirus along the L4-L5 dorsal root entry zone using a glass micropipette (diameter 60 μm). The tip of glass micropipette reached to the depth of lamina II-IV of the spinal cord. The dorsal muscle and skin were then sutured.
Behavioral analysis
Animals were habituated to the testing environment daily for 2 days before baseline testing. All the behavioral experiments were done by individuals blinded to the treatment of the mice. For heat hyperalgesia, the animals were put in a plastic box placed on a glass plate, and the plantar surface was exposed to a beam of radiant heat through a transparent glass surface (IITC model 390 Analgesia Meter, Life Science). The baseline latencies were adjusted to 10–14 s with a maximum of 20 s as cutoff to prevent potential injury. 23 For mechanical allodynia, the animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of the hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 grams, Stoelting, Wood Dale, IL), presented perpendicular to the plantar surface. The 50% paw withdrawal threshold was determined using Dixon’s up-down method.
Spinal slice preparation
The lumbar spinal cord was carefully removed from mice (4–6 weeks) under urethane anesthesia (1.5–2 g/kg, i.p.) and placed in preoxygenated (saturated with 95% O2 and 5% CO2) ice-cold sucrose artificial CSF (aCSF) solution. The sucrose aCSF contains the following (in mM): 234 sucrose, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 12 glucose, and 25 NaHCO3. The pia-arachnoid membrane was gently removed from the section. The portion of the lumbar spinal cord (L4–L5) was identified by the lumbar enlargement and large dorsal roots. The spinal segment was placed in a shallow groove formed in an agar block and then glued to the button stage of a VT1000S vibratome (Leica). Transverse slices (450 μm) were cut in the ice-cold sucrose aCSF, incubated in Krebs’ solution oxygenated with 95% O2 and 5% CO2 at 34°C for 30 min, and then allowed to recover 1–2 h at room temperature before the experiment. The Krebs’ solution contains the following (in mM): 117 NaCl, 3.6 KCl, 1.2 MgCl2, 2.5CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose.
Patch-clamp recordings in spinal cord slices
The voltage-clamp recordings were made from neurons in outer lamina II of the dorsal horn. The slice was continuously superfused (3–5 ml/min) with Krebs’ solution in room temperature, and saturated with 95% O2 and 5% CO2. Individual neurons were visualized under a stage-fixed upright infrared differential interference contrast microscope (BX51WI, Olympus) equipped with a 40 water-immersion objective. The patch pipettes were pulled using a Flaming micropipette puller (P-97, Sutter Instruments), and had initial resistance of 5–10 M when filled with the internal pipette solution contained the following (in mM): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, and 5 Na2ATP. Membrane voltage and current were amplified with a multiclamp 700B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 10 kHz using a data acquisition interface (1440A, Molecular Devices). A seal resistance (>2 GΩ) and an access resistance (<35 MΩ) were considered acceptable. The cell capacity transients were cancelled by the capacitive cancellation circuitry on the amplifier. After establishing the whole-cell configuration, the membrane potential was held at −70 mV for recording sEPSC and mEPSC and at 0 mV for sIPSC. Data were stored with a personal computer using pClamp10.0 software and analyzed with Mini Analysis (Synaptosoft 6.0). Those cells that showed >10% changes from the baseline levels were regarded as responsive to the presence of drugs.
Quantification and statistics
All data were expressed as mean ± SEM. The behavioral data were analyzed by two-way repeated-measures (RM) ANOVA followed by Bonferroni’s test as the post hoc multiple comparison analysis. For Western blot, the density of specific bands was measured with ImageJ. The levels of CXCL11 and pERK were normalized to loading control GAPDH. Student’s t test was applied when only two groups needed to be compared. The criterion for statistical significance was p < 0.05.
Results
CXCL9 expression is upregulated in spinal astrocytes after SNL
Previous report has shown that SNL induces persistent (> 21 days) mechanical allodynia and heat hyperalgesia. 13 We examined the time course of Cxcl9 expression in the spinal cord at days 1, 3, 10, and 21 after SNL. SNL induced persistent Cxcl9 mRNA upregulation, which started at day 3, peaked at day 10, and was still elevated at day 21 (p < 0.05 or 0.01, SNL vs. Sham, Figure 1(a)). CXCL9 protein level was also significantly increased 10 days after SNL (p < 0.05, Figure 1(b)). Immunostaining revealed basal expression of CXCL9 in the dorsal horn in naive (Figure 1(c)) and sham mice (data not shown), but markedly increased in the dorsal horn 10 days after SNL (Figure 1(d)). In addition, preabsorption of CXCL9 antibody with the CXCL9 blocking peptide abolished the immunostaining signal in the spinal cord (Figure 1(e)).

The CXCL9 expression is increased in spinal astrocytes after SNL. (a) Time course of Cxcl9 mRNA expression in the ipsilateral dorsal horn in naive, sham-operated, and SNL mice. Cxcl9 expression was significantly increased at 3, 10, and 21 days in SNL mice. *p < 0.05, **p < 0.01, compared with sham-operated mice. Student's t test. n = 5 mice/group. (b) ELISA shows the increase of CXCL9 protein in the spinal cord 10 days after SNL. *p < 0.05, compared with sham-operated mice. Student’s t test, n = 5 mice/group. (c to d) Representative images of CXCL9 immunofluorescence in the spinal cord from naïve and SNL mice, receptively. CXCL9 was constitutively expressed in naive mice (c), but significantly increased in the ipsilateral dorsal horn 10 days after SNL mice (d). (e) CXCL9-IR was not shown after absorption with CXCL9 peptide. (f to h) Double staining shows the cellular distribution of CXCL9 in the spinal dorsal horn. CXCL9 was sparely colocalized with NeuN (f) or CD11b (g), but highly colocalized with GFAP (h) in the spinal cord 10 days after SNL.
To define the cellular localization of CXCL9 in the spinal cord, we further did double staining. At SNL day 10, CXCL9 was sparely colocalized with neuronal marker NeuN (Figure 1(f)) or microglial marker CD11b (Figure 1(g)), but highly colocalized with astrocytic marker GFAP (Figure 1(h)). These results suggest that CXCL9 was increased in the dorsal horn and mainly expressed in astrocytes after SNL.
CXCL11 is upregulated in spinal astrocytes after SNL
We then examined the expression of CXCL11 in the spinal cord after SNL or sham operation. As shown in Figure 2(a), Cxcl11 mRNA was markedly increased at day 1, day 3, peaked at day 10, and was still upregulated at day 21 in SNL mice compared to sham-operated mice (p < 0.01 or 0.001, SNL vs. Sham). To detect the protein level of CXCL11, we used Western blot instead of ELISA, as the ELISA kit for CXCL11 was not commercially available. As shown in Figure 2(b), CXCL11 protein was also significantly increased 10 days after SNL (p < 0.05). Immunostaining revealed low expression of CXCL11 in the dorsal horn in naive (Figure 2(c)) and sham-operated mice (data not shown), but increased expression in the dorsal horn 10 days after SNL (Figure 2(d)). In addition, CXCL11 blocking peptide abolished the immunostaining signal of CXCL11 antibody in the spinal cord (Figure 2(e)).
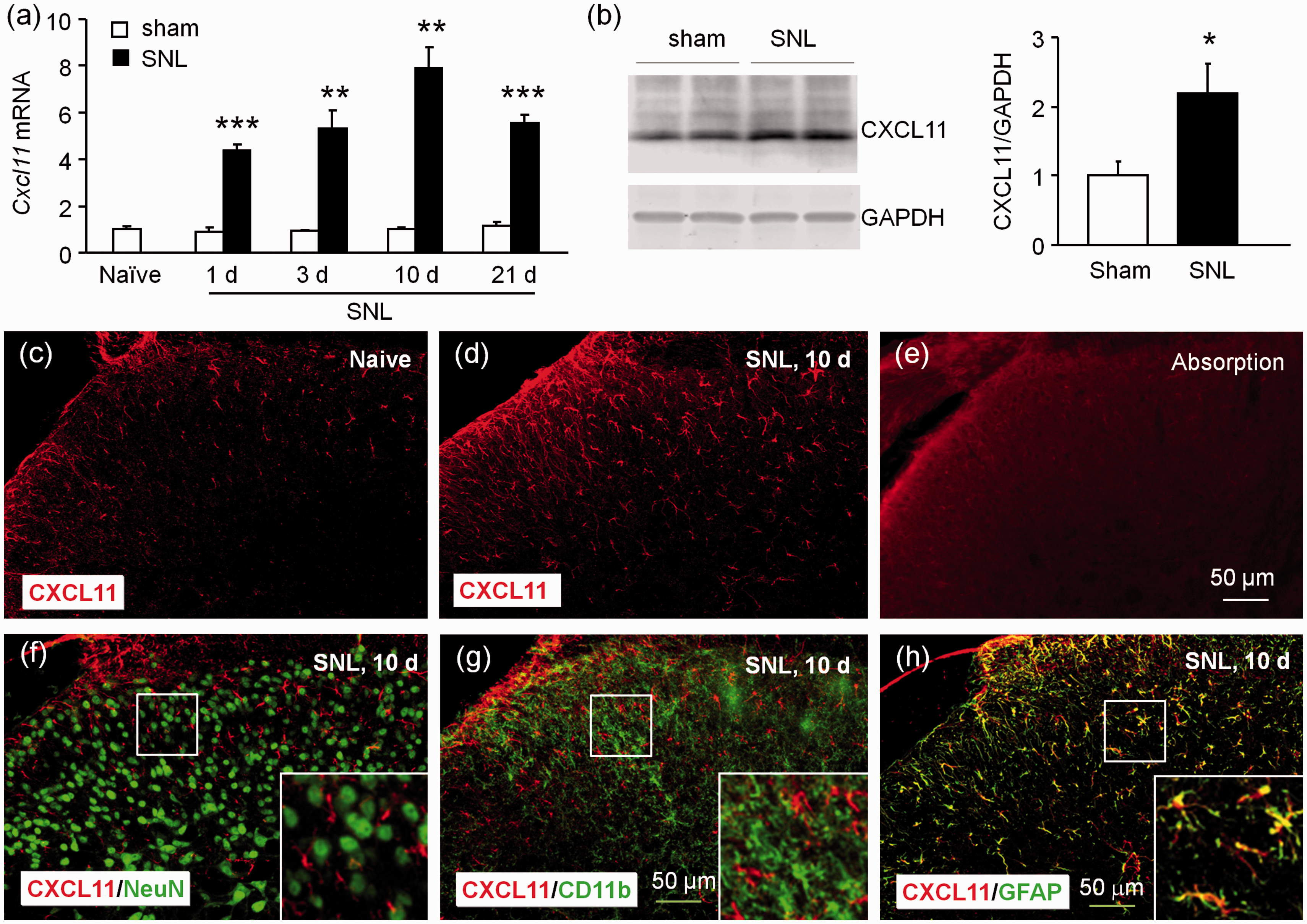
CXCL11 expression is increased in spinal astrocytes after SNL. (a) Time course of Cxcl11 mRNA expression in the ipsilateral dorsal horn in naive, sham-operated, and SNL mice. Cxcl11 expression was increased at 1, 3, 10, and 21 days in SNL mice, compared to in sham-operated mice. **p < 0.01, ***p < 0.001, Student’s t test. n = 5 mice/group. (b) Western blot shows the increase of CXCL11 protein in the spinal cord 10 days after SNL. *p < 0.05, Student’s t test, n = 4 mice/group. (c to d) Representative images of CXCL11 immunofluorescence in the spinal cord from sham and SNL mice, receptively. CXCL11 was constitutively expressed in naive (c) mice, but markedly increased in the ipsilateral dorsal horn 10 days after SNL (d). (e) CXCL11-IR was not shown after absorption with CXCL11 peptide. (f to h) Double staining shows the cellular distribution of CXCL11 in the spinal dorsal horn. CXCL11 was not double-stained with NeuN (f) or CD11b (g), but highly colocalized with GFAP (h) in the spinal cord 10 days after SNL.
We then examined the distribution of CXCL11 in the spinal horn by double staining. Similar as CXCL9, CXCL11 was not colocalized with NeuN (Figure 2(f)) or CD11b (Figure 2(g)), but highly colocalized with GFAP (Figure 2(h)). These results suggest that CXCL11 was markedly increased in spinal astrocytes after SNL.
Inhibition of spinal CXCL9 or CXCL11 does not alleviate SNL-induced neuropathic pain
To examine whether CXCL9 and CXCL11 are involved in the pathogenesis of neuropathic pain, we prepared the recombinant lentivirus containing shRNA targeting Cxcl9, Cxcl11, or Cxcl10. In vitro experiment showed that all the three shRNA effectively reduced the expression of the corresponding chemokine (p < 0.05, Figure 3(a) to (c)). The lentivirus was intraspinally injected three days after SNL. As shown in Figure 3(d) to (e), intraspinal injection of LV-Cxcl9 shRNA or LV-Cxcl11 shRNA did not affect the paw withdrawal latency (Figure 3(d)) or threshold (p > 0.05, two-way RM ANOVA, Figure 3(e)) at days 10 and 14 after SNL. In comparison, LV-Cxcl10 shRNA markedly attenuated SNL-induced heat hyperalgesia (p < 0.001, two-way RM ANOVA, Figure 3(f)) and mechanical allodynia (p < 0.01, two-way RM ANOVA, Figure 3(g)) at days 10 and 14. These data suggest that CXCL9 and CXCL11 are not necessary for the maintenance of neuropathic pain.

Inhibition of CXCL9 or CXCL11 does not alleviate SNL-induced neuropathic pain. (a to c) The shRNA targeting Cxcl9, Cxcl11, or Cxcl10 reduced the mRNA expression of Cxcl9 (a), Cxcl11 (b), and Cxcl10 (c) in HEK293 cells. (d to e) Intraspinal injection of LV-Cxcl9 shRNA or LV-Cxcl11 shRNA three days after SNL did not change the paw withdrawal latency (d) or threshold (e) at days 10 and 14. p > 0.05, two-way RM ANOVA. n = 6–7 mice/group. (f to g) Intraspinal injection of LV-Cxcl10 shRNA three days after SNL significantly increased the paw withdrawal latency (f) and threshold (g) at days 10 and 14. ** p < 0.01, *** p <0.001, two-way RM ANOVA followed by Bonferroni’s test, n = 5–6 mice/group.
CXCL9 or CXCL11 does not induce pain hypersensitivity or ERK activation in the spinal cord
Previous studies have shown that intrathecal injection of chemokines, including CCL2, CXCL1, CXCL10, and CXCL13, induce robust pain hypersensitivity.11–13,21 We intrathecally injected the same dose of CXCL9 (100 ng) or CXCL11 (100 ng) and checked heat hyperalgesia and mechanical allodynia. The data showed that neither CXCL9 nor CXCL11 changed the paw withdrawal latency (p > 0.05, two-way RM ANOVA, Figure 4(a)) or paw withdrawal threshold in 6 h we tested (p > 0.05, two-way RM ANOVA, Figure 4(b)). We also injected much higher dose (200 ng) of CXCL9 or CXCL11, but the latency and threshold were not affected either (data not shown). In contrast, CXCL10 at 100 ng induced hyperalgesia at 1 h, 3 h, and 6 h (p < 0.001, two-way RM ANOVA, Figure 4(c)). CXCL10 also induced mechanical allodynia in 3 h (p < 0.001, two-way RM ANOVA, Figure 4(d)).
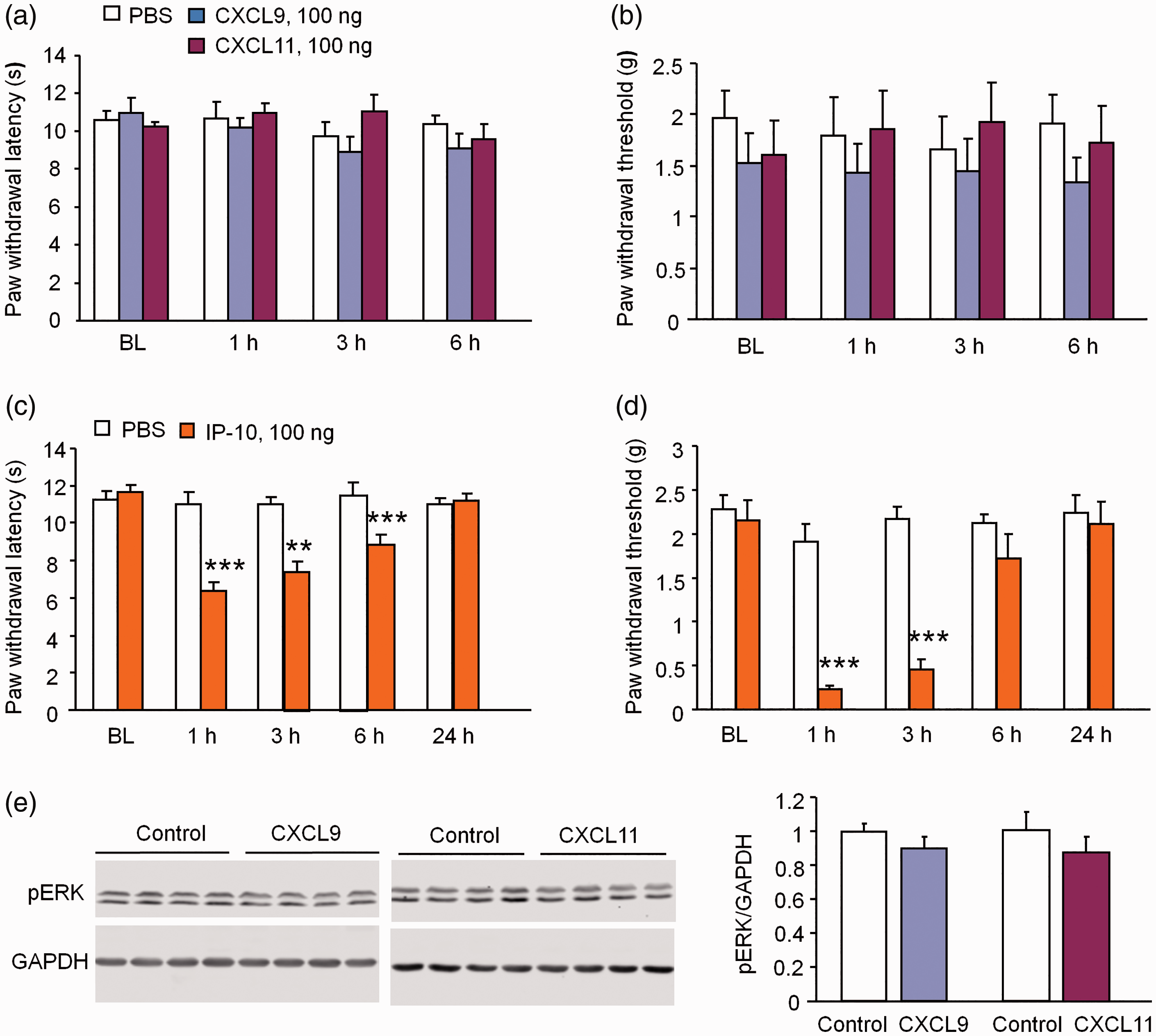
CXCL9 and CXCL11 do not induce pain hypersensitivity after intrathecal injection. (a, b) Intrathecal injection of CXCL9 and CXCL11 (100 ng) did not induce heat hyperalgesia (a) or mechanical allodynia (b) in naive mice. p > 0.05, two-way RM ANOVA. n = 6 mice/group. (c, d) Intrathecal injection of CXCL10 (100 ng) induced heat hyperalgesia (C) at 1, 3, and 6 h and mechanical allodynia (d) at 1 h and 3 h in naive mice. *** p < 0.001, two-way RM ANOVA followed by Bonferroni’s test, n = 6–7 mice/group. (e) pERK expression in the spinal cord did not change 1 h after intrathecal injection of CXCL9 or CXCL11 in naive mice. p > 0.05, Student's t test, n = 4 mice/group.
Activation of ERK in spinal horn neurons could serve as a marker for central sensitization. 24 CXCL10 induces pain hypersensitivity and ERK activation in the spinal cord. 21 To examine the activation of ERK after injection of CXCL9 or CXCL11, we then checked pERK expression in the spinal cord by Western blot. As shown in Figure 4(e), the pERK expression was not significantly changed 1 h after CXCL9 or CXCL11 injection (Figure 4(e)). These results suggest that CXCL9 and CXCL11 are not sufficient to induce pain hypersensitivity and central sensitization in naïve mice.
CXCL9 and CXCL11 increase the frequency of sEPSCs and mEPSCs in naïve mice
Since CXCL10 increases the excitatory synaptic transmission in lamina II neurons via CXCR3, 21 we asked whether CXCL9 and CXCL11 regulate synaptic transmission. We prepared spinal cord slices and performed whole cell patch-clamp recordings in spinal horn neurons in naive mice. We first recorded sEPSCs in lamina II neurons. Superfusion of CXCL9 (100 ng/ml) significantly increased the frequency of sEPSCs in 15 of 22 neurons (68.2%) recorded from naive mice (Figure 5(a)). However, the amplitude of sEPSCs was not changed (Figure 5(a)). The same concentration of CXCL11 also induced a similar increase of the frequency of sEPSCs in 13 of 19 neurons (68.4%), but did not affect the amplitude (Figure 5(b)).

CXCL9 and CXCL11 enhance excitatory synaptic transmission in lamina II neurons. (a) Whole-cell patch clamp recording of sEPSCs shows an increase in the frequency of sEPSCs after perfusion of CXCL9 (100 ng/ml, 2 min). However, the amplitude was not changed after perfusion of CXCL9. a1 and a2 are enlarged traces before and after CXCL9 treatment, respectively. ***p < 0.001 versus pretreatment baseline, Student’s t test, n = 5–6 mice/group. (b) sEPSCs frequency not amplitude is increased after perfusion of CXCL11 (100 ng/ml, 2 min). b1 and b2 are enlarged traces before and after CXCL11 treatment, respectively. **p < 0.01 versus pretreatment baseline, Student’s t test, n = 5–6 mice/group. (c) Miniature EPSCs (mEPSCs) were recorded in the present of TTX (500 nM). The mEPSCs frequency, not the amplitude is increased after perfusion of CXCL9 (100 ng/ml, 2 min). c1 and c2 are enlarged traces before and after CXCL9 treatment, respectively. *p < 0.05 versus pretreatment baseline, Student’s t test, n = 3 mice/group. (d) mEPSCs frequency, not the amplitude is increased after incubation of CXCL11. d1 and d2 are enlarged traces before and after CXCL11 treatment, respectively. * p < 0.05 versus pretreatment baseline, Student's t test, n = 3 mice/group.
We further checked the influence of CXCL9 and CXCL11 on mESPCs in lamina II neurons, and 500 nM TTX was added in the aCSF. As shown in Figure 5(c), superfusion of CXCL9 (100 ng/ml) significantly increased the frequency of mEPSCs in 8 of 10 neurons (80%) recorded from naive mice (p < 0.05, Student’s t test), but did not change the amplitude. Superfusion of CXCL11 (100 ng/ml) also significantly increased the frequency, but not amplitude of mEPSCs in 7 of 8 neurons (87.5%) recorded in experiments (p < 0.05, Student’s t test, Figure 5(d)).
CXCL9 and CXCL11 increase the frequency of sIPSC in naïve mice
Spinal disinhibition, such as loss of GABAergic inhibition, is thought to be one of the essential mechanisms of neuropathic pain. 16 Previous study showed that proinflammatory cytokines such as IL-1β not only increases the excitatory synaptic transmission but also decreases inhibitory synaptic transmission in the spinal cord. 25 To investigate the effect of CXCL9 and CXCL11 on the inhibitory synaptic transmission, we examined the sIPSCs in lamina II neurons. As shown in Figure 6(a), the frequency, but not the amplitude of sIPSCs, was markedly increased by CXCL9 in 91.7% (11/12) recorded neurons (p < 0.05, Student’s t test). Additionally, CXCL11 also increased frequency of the sIPSCs in 81.8% (9/11) recorded neurons (p < 0.001, Student’s t test, Figure 6(b)). As a comparison, CXCL10 (100 ng/ml) did not significantly change the frequency or the amplitude of sIPSCs in 10 recorded neurons (p > 0.05, Student’s t test, Figure 6(c)). These results suggest that CXCL9 and CXCL11 enhance the frequency of both sEPSCs and sIPSCs, but CXCL10 did not change the inhibitory synaptic transmission in spinal horn neurons.

CXCL9 and CXCL11 enhance inhibitory synaptic transmission in lamina II neurons. (a) Patch clamp recording shows an increase in the frequency but not the amplitude of the sIPSC after perfusion of CXCL9 (100 ng/ml, 2 min). a1 and a2 are enlarged traces before and after CXCL9 treatment. *p < 0.05, Student’s t test, n = 3–4 mice/group. (b) The frequency, not the amplitude of the sIPSCs was increased during perfusion of CXCL11 (100 ng/ml, 2 min). b1 and b2 are enlarged traces before and after CXCL11 treatment. ***p < 0.001, Student’s t test, n = 3–4 mice/group. (c) Neither the frequency nor the amplitude of the sIPSCs was changed after perfusion of CXCL10 (100 ng/ml, 2 min). c1 and c2 are enlarged traces before and after CXCL10 treatment. p > 0.05, Student’s t test, n = 3–4 mice/group.
Discussion
In this study, we show that CXCR3 ligands, CXCL9 and CXCL11, similar as CXCL10, were upregulated in spinal astrocytes after SNL. However, CXCL9 and CXCL11 play different roles from CXCL10 in mediating neuropathic pain. Inhibition of CXCL9 or CXCL11 did not attenuate neuropathic pain, whereas inhibition of CXCL10 markedly alleviated SNL-induced heat hyperalgesia and mechanical allodynia. Furthermore, intrathecal injection of CXCL9 and CXCL11 did not induce pain hypersensitivity, but CXCL10 did. Consistently, spinal ERK was not activated after injection of CXCL9 and CXCL11. Our electrophysiological recording results further demonstrated that CXCL9 and CXCL11 enhanced both excitatory and inhibitory synaptic transmission, but CXCL10 only enhanced excitatory synaptic transmission in dorsal horn neurons.
The chemokines CXCL9, CXCL10, and CXCL11 as the non-ELR CXC chemokine subgroup are strongly induced by infection, allograft rejection, injury, or immunoinflammatory responses. 26 Previous studies have reported that these chemokines are detectable in peripheral blood leukocytes, liver, thymus, spleen, lung, and are also expressed in astrocytes and microglia in central nervous system.4,27,28 Here, we observed that, after SNL, the expression of spinal CXCL9 and CXCL11 were both markedly upregulated, but CXCL11 is upregulated earlier than CXCL9 or other chemokines including CXCL10, 21 CXCL1, 13 and CCL211 in mice after SNL. In addition, CXCL9 was constitutively expressed in both neurons and glial cells, but only increased in astrocytes after SNL, whereas CXCL11 was expressed and increased in astrocytes. Our previous study showed that CXCL10 was expressed mainly in neurons in naïve mice, and markedly upregulated in astrocytes in SNL mice. 21 It was reported that CXCL10 and CXCL11 are also predominantly expressed by astrocytes in the spinal cord of animals with experimental autoimmune encephalomyelitis.29,30 Miu et al. 31 detected CXCL9 expression in microglia and CXCL10 expression in astrocytes in brain by in situ hybridization, suggesting different cellular distribution in different areas.
CXCR3 has been demonstrated to play a pivotal role in the development and maintenance of chronic pain.21,32 Cxcr3 deficient mice showed reduced hyperalgesia and allodynia after SNL. In addition, inhibition of CXCR3 by specific inhibitor or shRNA attenuated SNL-induced pain hypersensitivity. 21 Previous studies showed that inhibition of spinal chemokines including CCL2, CXCL1, and CXCL13 alleviated neuropathic pain.11–13 Here, inhibition of CXCL10 by shRNA attenuated SNL-induced heat hyperalgesia and mechanical allodynia, whereas inhibition of CXCL9 or CXCL11 by the same amount of shRNA did not show any effect. These data suggest that CXCL10, but not CXCL9 or CXCL11, is the major receptor for the activation of CXCR3.
Behavioral studies also showed that intrathecal injection of CXCL10 induced thermal and mechanical hypersensitivity, whereas CXCL9 and CXCL11 did not induce a similar hyperalgesia or allodynia. In addition, ERK activation in dorsal horn neurons contributes importantly to the induction of central sensitization.33,34 However, CXCL9 or CXCL11 application to the spinal cord did not induce the up-regulation of pERK, which is different from the effect of CXCL10, 21 suggesting that CXCL9 and CXCL11 are not sufficient to induce central sensitization and pain hypersensitivity.
Hyperactivity of excitatory synaptic transmission is one of the essential mechanisms for the induction of central sensitization. 35 Previous studies have demonstrated that persistent perfusion with chemokines including CXCL10, 21 CXCL1, 36 and CCL211 induce a significant enhancement of excitatory synaptic transmission in the spinal cord. Consistent with this phenomenon, we also found that the excitatory synaptic transmission in lamina II neurons was markedly increased during CXCL9 or CXCL11 treatment. The γ-aminobutyric acid (GABA) or glycine inhibit the synaptic transmission of noxious sensory signals in the spinal cord and previous studies have demonstrated that spinal GABAergic inhibition on synaptic transmission is reduced during neuropathic pain. 16 However, the frequency of inhibitory synaptic transmission from spinal cord was increased during CXCL9 or CXCL11 perfusion. The enhancement of frequency of sIPSCs in the spinal cord by CXCL9 and CXCL11 may antagonize the increase of glutamatergic synapses sensitization, thus no pain hypersensitivity was shown after intrathecal injection. The detailed mechanisms under these changes need further investigations in the future.
Previous studies have implicated that CXCL9, CXCL10, and CXCL11 may work redundantly, collaboratively, or antagonistically. 26 Although all of them can bind and activate CXCR3, distinct intracellular domains are required: CXCL9 and CXCL10 require the carboxy-terminal domain and beta-arrestin-1 binding domain, whereas CXCL11 requires the third intracellular loop, 37 which may cause difference in the downstream responses. In addition, for all three ligands, the affinity to CXCR3 is different. In vitro studies showed that CXCL11 has the highest affinity binding to CXCR3, followed by CXCL10 having intermediate affinity and then CXCL9 having the lowest affinity.4,38,39 Furthermore, CXCR3 may not be the only receptor of these chemokines. For example, CXCL11 also bind CXCR7. 40 Whether these differences among the three ligands contribute to the different role of CXCL9/CXCL11 and CXCL10 in pain hypersensitivity needs to be further investigated.
In summary, our results demonstrate the distinct roles of chemokines CXCL9, CXCL10, and CXCL11 in pain hypersensitivity. Although CXCL9 and CXCL11 are upregulated after nerve injury, they may not contribute to the pathogenesis of neuropathic pain. CXCL9 and CXCL11 also showed different role from CXCL10 in mediating inhibitory synaptic transmissions. These data suggest that CXCL9 and CXCL11 may not be the effective targets in the treatment of neuropathic pain.
Footnotes
Authors’ Contribution
XBW carried out electrophysiological recording experiments, LNH performed the animal surgery, behavioral testing, immunohistochemistry, and Western blot experiments. BCJ, HS, XQB, and WWZ performed the PCR, immunohistochemistry, and behavioral experiments. YJG conceived of the project, coordinated and supervised the experiments. XBW and YJG wrote the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (NSFC 31671091 and 81771197), the Natural Science Foundation of Jiangsu Province (BK20171255), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.