
Abstract
Background
Nociceptive and neuropathic pain occurs as part of the disease process after traumatic brain injury (TBI) in humans. Central and peripheral inflammation, a major secondary injury process initiated by the traumatic brain injury event, has been implicated in the potentiation of peripheral nociceptive pain. We hypothesized that the inflammatory response to diffuse traumatic brain injury potentiates persistent pain through prolonged immune dysregulation.
Results
To test this, adult, male C57BL/6 mice were subjected to midline fluid percussion brain injury or to sham procedure. One cohort of mice was analyzed for inflammation-related cytokine levels in cortical biopsies and serum along an acute time course. In a second cohort, peripheral inflammation was induced seven days after surgery/injury with an intraplantar injection of carrageenan. This was followed by measurement of mechanical hyperalgesia, glial fibrillary acidic protein and Iba1 immunohistochemical analysis of neuroinflammation in the brain, and flow cytometric analysis of T-cell differentiation in mucosal lymph. Traumatic brain injury increased interleukin-6 and chemokine ligand 1 levels in the cortex and serum that peaked within 1–9 h and then resolved. Intraplantar carrageenan produced mechanical hyperalgesia that was potentiated by traumatic brain injury. Further, mucosal T cells from brain-injured mice showed a distinct deficiency in the ability to differentiate into inflammation-suppressing regulatory T cells (Tregs).
Conclusions
We conclude that traumatic brain injury increased the inflammatory pain associated with cutaneous inflammation by contributing to systemic immune dysregulation. Regulatory T cells are immune suppressors and failure of T cells to differentiate into regulatory T cells leads to unregulated cytokine production which may contribute to the potentiation of peripheral pain through the excitation of peripheral sensory neurons. In addition, regulatory T cells are identified as a potential target for therapeutic rebalancing of peripheral immune homeostasis to improve functional outcome and decrease the incidence of peripheral inflammatory pain following traumatic brain injury.
Background
Traumatic brain injury (TBI) is a major cause of death and disability throughout the world, with few treatments for those who suffer from subsequent lifelong neurological symptoms. The somatic symptoms of TBI include sensory hypersensitivity and persistent pain, which encompasses headache, nociceptive pain, and neuropathic pain, the latter of which is defined as pain initiated or caused by a lesion or disease of the somatosensory system. Although epidemiological data support the occurrence of pain as a secondary complication of TBI,1–5 only a limited description of postinjury pain in experimental models is available, 6 with even fewer investigations of underlying mechanisms. 7
For the intact nervous system, a stimulus of sufficient magnitude to evoke the sensation of pain can directly damage tissue and this serves an adaptive function; 8 however, under pathological conditions, sensitization can occur resulting in maladaptive pain states.9,10 In these instances, tissue injury causes changes in neuronal activation of pain pathways, thus increasing the excitability of central nervous system (CNS) neurons.9–11 Together, these neuronal changes result in sensitization associated with evoked hypersensitivity to typically painful (hyperalgesia) and nonpainful (allodynia) stimuli.9,12,13
TBI results in neuronal cell death, ischemia, hemorrhage, and the disruption of the blood–brain barrier (BBB). These primary insults initiate a secondary injury response of cellular and molecular cascades including central inflammation and the production of cytokines. 14 Furthermore, TBI-induced disruption of the BBB allows passage of inflammatory cytokines out of the injured brain, thereby initiating a systemic immune response. 15 Together, these central and peripheral immune responses contribute to microglia activation, astrocytosis, leukocytic infiltration, 16 and the secretion of immunological factors and pronociceptive mediators (e.g. cytokines). The overall increase in inflammation-mediating cytokines and chemokines establish neuronal hypersensitivity,10,11,13,17 which we hypothesize induces hyperalgesia.9–11,13,17,18 In addition, the persistent inflammatory signaling maintain immune cells in an activated state, such that they are primed for future challenges and insults. This primed profile may set the stage for hyperactivity to challenges including a peripheral immune response (for review, see Witcher et al.). 19
The composition of cytokines and regulatory factors in the surrounding environment determine the functional lineage in mature T cells as either proinflammatory effector helper T cells (Th) or anti-inflammatory T-regulatory cells (Tregs).
20
Tregs contribute to maintaining immune cell homeostasis
21
by controlling not only the specificity and intensity of effector T cell function but also the proinflammatory reactivity of innate immune cells (see Figure 1).22,23 For example, Tregs originating in the thymus can prevent autoimmune reactions, while adaptive (induced) Tregs have a more prominent anti-inflammatory activity and are instrumental in the control of inflammation-associated immune reactions.24–26 In this context, previous studies suggested that Tregs contribute to the resolution of brain-injury-induced inflammation.
12
Experimentally, mouse Tregs can be identified by the expression of the forkhead family transcription factor (Foxp3) in combination with the high expression of the interleukin 2 (IL-2) receptor alpha subunit, CD25.
Proposed mechanistic link between unregulated peripheral inflammation and inflammatory hyperalgesia following TBI. (a) In uninjured sham mice, carrageenan injection into the paw (1) initiates a pro-inflammatory immune response that is homeostatically regulated (2). Anti-inflammatory actions of Tregs suppress inflammation and result in a normal response to painful stimuli in the von Frey filament test (3). (b) Diffuse brain injury induces systemic immune dysregulation, specifically through deficiencies in T cell proliferation and differentiation of Tregs (1). Carrageenan injection into the paw (2) initiates an inflammatory response that is unregulated in the absence of Tregs (3). Resulting proinflammatory cytokines cause aberrant excitation of sensory neurons (4), potentiating hyperalgesia in the von Frey filament test (5).
Here, we hypothesize that TBI potentiates hyperalgesia by dysregulating the T cell response and increasing the susceptibility (priming) of the systemic immune system to respond to a secondary challenge or insult. Following midline fluid percussion injury (FPI), inflammation and hyperalgesia were tested after carrageenan injection in the hind paw. The central immune response to diffuse TBI was assessed by microglia and astrocyte0 morphology and inflammatory mediator levels in the cortex. The peripheral immune response was assayed by inflammatory mediator levels in the serum. Finally, the response of mucosal lymph node T cells to all-trans retinoic acid (ATRA), a potent inducer of Tregs, was tested to evaluate T cell differentiation following TBI. Results indicated that diffuse brain injury increased inflammatory pain associated with cutaneous inflammation by priming the systemic immune system toward a proinflammatory response. Furthermore, T cell dysfunction may have contributed to unresolved peripheral inflammation which potentiated mechanical hyperalgesia (see Figure 1).
Methods
Animals
Male C57BL/6 mice (Harlan Laboratories, Inc., Indianapolis, IN) were used for all experiments. The animals were housed at a constant temperature (23℃ ± 2℃) with food and water available ad libitum according to the Association for Assessment and Accreditation of Laboratory Animal Care International. Animals were acclimated to their environment following shipment for at least three days prior to any experiments. After surgery, postoperative care included daily evaluation and documentation of the health status of each animal with a physical examination. Animal care and procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Midline fluid percussion injury
Adult male C57BL/6 mice (20–24 g) were subjected to midline fluid percussion injury (mFPI) consistent with methods previously described. 27 Mice were anesthetized using 5% isoflurane in 100% oxygen for 5 min and the head of the animal was placed in a stereotaxic frame with continuously delivered isoflurane at 2.5% via nosecone. While anesthetized, body temperature was maintained using a Deltaphase isothermal heating pad (Braintree Scientific Inc., Braintree, MA). A midline incision was made exposing bregma and lambda, and fascia was removed from the surface of the skull. A midline craniotomy was performed via trephination (3 mm outer diameter) between the bregma and lambda joints where a modified Luer lock hub was affixed using cyanoacrylate gel and methyl-methacrylate (Hygenic Corp., Akron, OH) over the exposed dura. The injury cap was closed using a Luer lock cap and animals were placed in a heated recovery cage and monitored until ambulatory before being returned to their cage.
For injury induction 24 hours postsurgery, animals were reanesthetized with 5% isoflurane delivered for 5 min. The cap was removed from the injury-hub assembly and the craniotomy was visually inspected through the hub. The hub was then filled with normal saline and attached to the male end of the fluid percussion device (Custom Design and Fabrication, Virginia Commonwealth University, Richmond, VA). Pendulum release resulted in a diffuse brain injury (average of 1.4 atm). Sham animals were attached to and removed from the fluid percussion device without dropping the pendulum. Animals were monitored for righting reflex recovery time and for the presence of a forearm fencing response as a metric of injury severity. 28 The righting reflex time is the total time from the initial impact until the animal spontaneously rights itself. The fencing response is a tonic posturing characterized by extension and flexion of opposite arms that has been validated as an overt indicator of injury severity. 28 The injury hub was removed and the brain was inspected for uniform herniation and integrity of the dura. After spontaneously righting, animals were placed in a heated recovery cage and monitored until ambulatory before being returned to their cage (approximately 5 to 15 min).
Tissue preparation and cytokine measurement
At selected time points (1, 3, 9, 12, 24, 48, and 168 h) postsurgery, mice were given an overdose of sodium pentobarbital and transcardially perfused with phosphate-buffered saline (PBS) after cardiac blood draws. Mice were decapitated and the brains were dissected on ice and samples were snap frozen in liquid nitrogen then stored at −80℃ until used. The protein levels of a panel of inflammatory cytokines were measured in the cortex and serum by Meso Scale Discovery (MSD) multiplex immunoassay (sector imager 2400, Meso Scale Discovery; Gaithersburg, MD) as previously described.29,30 A PBS soluble brain homogenate was prepared from cortical tissue using a high shear homogenizer (Omni TH115), in a 1:10 (w/v) of ice-cold, freshly prepared lysis buffer consisting of 1 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride, and 1 mM ethylenediaminetetraacetic acid. The cortical homogenate was centrifuged at 12,000 x g for 20 min at 4℃, and supernatants were collected. MSD custom multiplex high-sensitivity ELISA kits were used according to the manufacturer’s instructions with minor modifications. Briefly, 25μl of undiluted serum or 100 µg of protein of the PBS soluble brain supernatant was loaded per well of the MSD plate. The sample was incubated at 4℃ for ∼14 h. All incubation steps were done using an Eppendorf MixMate at 1000 rpm. Brain samples were normalized to the total amount of protein in the sample loaded as determined by BCA Protein Assay (Thermo Scientific, Waltham, MA, USA).
Mechanical hyperalgesia testing
All testing began between 10:00 am and 12:00 pm in a temperature- and light-controlled room. Mice were acclimated for 30–60 min in the testing environment within a custom rectangular plastic box (15 × 4 × 4 cm; three white opaque walls and one clear wall) on a raised metal mesh platform. Baseline testing was conducted at seven days after TBI, and then at 2 and 4 h after carrageenan injection (see below for methods). To evaluate mechanical hypersensitivity (hyperalgesia), we used a logarithmically increasing set of eight von Frey filaments (Stoelting, Illinois), ranging in gram force from 0.007 to 6.0 g, as previously described. 31 These were applied perpendicular to the ventral-medial hind paw surface with sufficient force to cause a slight bending of the filament. A positive response was characterized as a rapid withdrawal of the paw away from the stimulus filament within 4 s. Using the up-down statistical method (Chaplan et al., 1994), the 50% withdrawal mechanical threshold scores were calculated for each mouse. We then calculated the carrageenan-induced change in mechanical threshold as the maximum possible effect (MPE) for each individual mouse with the following formula: MPE = ([50% threshold after carrageenan − baseline 50% threshold]/baseline 50% threshold) × 100.
Carrageenan model of inflammatory pain
At seven days postinjury, a baseline assessment of mechanical sensitivity was obtained prior to mice being lightly restrained and injected with low-dose carrageenan (0.2% in 5 µl 0.9% saline) into the intraplantar surface (ventral-medial) of the left hind paw. 32 To control for needle puncture and injectate volume, a mock procedure consisted of an intraplantar injection of saline (5 µl, 0.9%).
Tissue preparation for histology
At seven days postinjury or sham operation, mice used for mechanical threshold testing were given an overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde after a PBS flush. Brains were removed and placed in 4% paraformaldehyde overnight then immersed in serial dilutions (10%, 20%, and 30%) of sucrose for 24 h each. The brains were removed from sucrose and frozen at −20℃. After freezing, brains were sectioned on a cryostat in the coronal plane at 20 µm, mounted onto glass slides, and stored at −80℃.
Immunofluorescence
Slides with brain sections were removed from −80℃, placed in an oven at 60℃ for approximately 4 h and then rinsed three times for 5 min each in PBS. Next, the slides were blocked in 4% v/v donkey serum in PBS. Primary antibodies were added to 1% blocking solution, rabbit anti-ionized calcium binding adaptor molecule 1 (Iba-1; 1:2000, WAKO, cat# 019919741) or rabbit anti-glial fibrillary acidic protein (1:5000, DAKO, cat# Z033429-2) and stored at 4℃ overnight. Slides were rinsed three times in PBS and the secondary antibody (donkey anti-rabbit Alexa Fluor 488 or 594) was added and slides were incubated on a rocker at room temperature for 1 h. Finally, slides were rinsed in deionized water and coverslipped with antifade medium (Fluoromount G; Southern Biotech, Birmingham, AL). The cortex was examined for microglia and astrocyte activation in response to brain injury using a Zeiss LSM 710 laser scanning confocal microscope with attached digital camera.
Induction of regulatory T cells
Mucosal lymph nodes were removed postmortem from brain-injured (n = 4) and sham (n = 4) mice seven days postinjury. Lymph node cells were then isolated via mechanical disruption and centrifugation. Cells were resuspended at a density of 2 × 106/mL in iTreg medium: RPMI-1640 GlutaMAX (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 50 μM β-mercaptoethanol (Sigma-Aldrich), 2 ng/mL TGF-β (eBioscience), and 1 ng/mL IL-2 (eBioscience). 500 µL (1 × 106 cells) were incubated in 48-well tissue culture plates precoated with 500 ng/mL α-CD3 (clone: 145–2C11, Bio X Cell) for six days at 37℃/5% CO2. Half of the medium was replaced on day 3 of culture. When indicated, 1 μM ATRA (Sigma-Aldrich, St. Louis, MO) or 0.5 mg/mL soluble α-CD28 (clone: 37.51, eBioscience, San Diego, CA) was added to the culture medium.
Flow cytometric analysis of Treg development
Cells were stained using Foxp3 Staining Buffer Set and the following antibodies from eBioscience: α-CD25-AF-488, α-CTLA-4-PE, α-CD4-PE-Cy5, and α-CD62L-PE-Cy5 or from Miltenyi: Foxp3-APC. Some lymphocytes were labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA) per million cells for 3 min at room temperature. Fluorescent lymph node cells were assessed on a LSRII (BD Biosciences, San Jose, CA, USA) flow cytometer and analyzed with FlowJo software (Tree Star Inc., Ashland, OR, USA). Tregs (CD25hi/Foxp3hi) cells are reported as a percentage of the CD4+ cells with side and forward scatter profiles consistent with live lymphocytes (cells were collected as described earlier; sham n = 4, injury sham = 4).
Statistical analysis
Data are shown as mean ± SEM and analyzed using statistical software (GraphPad-Prism 6). Differences in mechanical threshold were determined using a repeated measures analysis of variance (ANOVA) followed by a Sidak’s multiple comparisons test. Cortical cytokine levels were determined with a one-way ANOVA followed by Dunnett’s multiple comparison test. Statistical significance was assigned for p < .05.
Results
Diffuse TBI potentiated mechanical hyperalgesia
At seven days postinjury, mechanical pain thresholds in the hind paws were tested to establish a baseline (preinjection) response for each animal. Baseline mechanical sensory thresholds were comparable in uninjured (n = 4) and brain-injured (n = 4) mice (t6 = 0.1549, p = 0.8820; data not shown). To test the hypothesis that diffuse TBI potentiates inflammatory pain, carrageenan was injected in the hind paw. At 2 and 4 h postinjection, mechanical threshold was decreased in brain-injured, compared to uninjured sham mice, F(1, 6) = 11.90, p = 0.0136; Figure 2(a). Decreases were also observed in the noninjected paw, but this failed to reach significance, F(1, 6) = 2.927, p = .1380; Figure 2(b).
TBI potentiated mechanical hyperalgesia in response to a peripheral immune challenge. (a and b) Mechanical hyperalgesia was assessed at seven days post-TBI (baseline; dotted line) and then 2 h and 4 h following carrageenan injection in the paw. Graphs depict the mechanical stimulation required to elicit a withdrawal response after carrageenan injection, as a percent of baseline, where values below 100% indicate increased sensitivity. Measurements were taken for the (a) injected and (b) noninjected paw (n = 4 per group, mean ± SEM, *p < 0.05).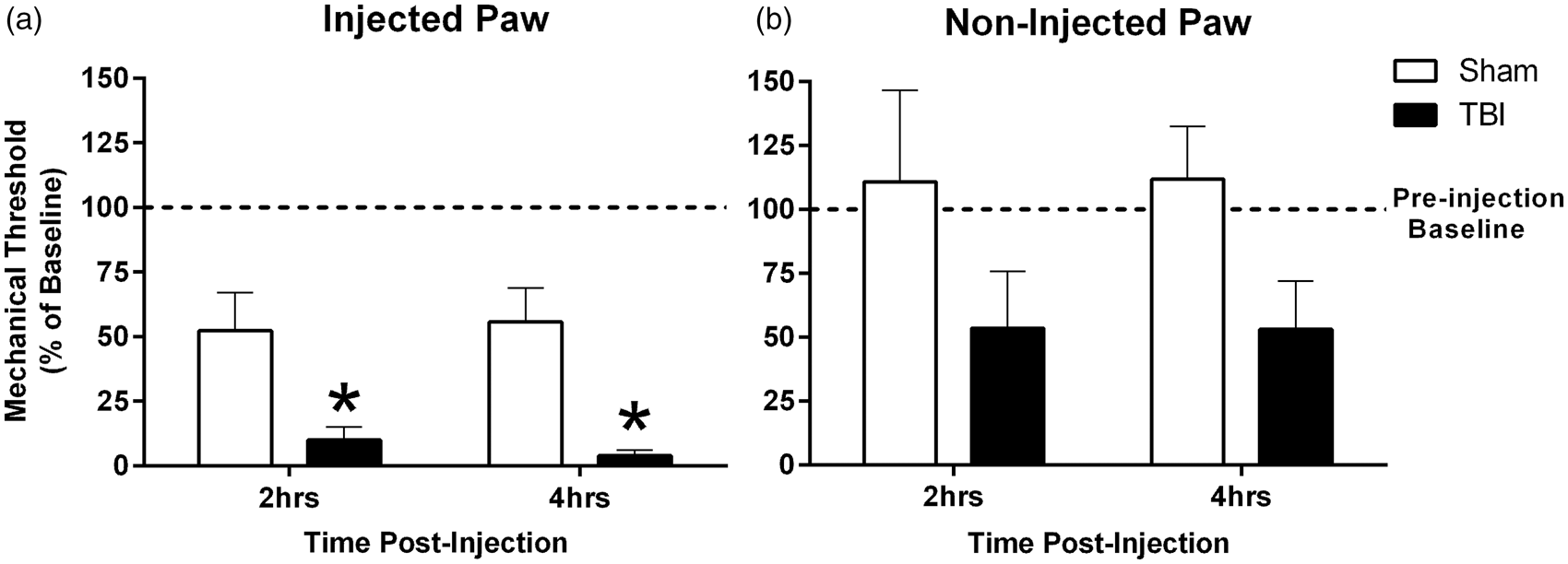
Diffuse TBI activated central and peripheral immune responses
IBA-1 immunohistochemistry identified microglia in the uninjured and brain-injured cortex. As previously reported by our group in the mFPI model, microglia in uninjured brain remain predominantly in a ramified (unactivated) morphology33,34 (Figure 3a). In the diffuse brain-injured cortex, microglia showed activated (rounded cell body with thickened processes) and amoeboid morphologies (Figure 3b), consistent with previous reports.33,34 In addition, activated astrocytes were found in brain-injured cortex (Figure 3d), indicated by hypertrophy, thickened processes, and enhanced expression of glial fibrillary acidic protein (GFAP), compared to sham (Figure 3c).
TBI activated microglia and astrocytes in the cortex. Seven days postinjury, cortical sections were immunostained for microglia and astrocyte activation using antibodies against (a and b) Iba-1 and (c and d) GFAP, respectively. (a) Highly ramified microglia from sham mice sharply contrasted with (b) activated microglia found following TBI. Increased process size was also noted among astrocytes from brain-injured mice (d) compared with uninjured sham (c). (40 × magnification, scale bars = 20 µm).
With a multiplex high-sensitivity ELISA, we found that diffuse brain injury led to rapid elevation in cortical protein levels of inflammatory cytokines (Figure 4a–d). Cortical IL-6 levels were significantly different between the uninjured sham and the brain injury time points, F(7, 19) = 2.779, p = 0.0361; Figure 4a), where Dunnett’s multiple comparison test indicated a significant increase in IL-6 at 9 h postinjury compared to uninjured sham. Cortical chemokine ligand 1 (CXCL1) levels were significantly different between uninjured sham and the brain injury time points, F(7, 19) = 1.617, p < 0.0001; Figure 4b), where Dunnett’s multiple comparison test indicated a significant increase in CXCL1 at 1, 3, and 9 h postinjury compared to uninjured sham. While there were injury-induced, immediate increases in cortical protein levels of TNF-α, F(7, 19) = 1.145, p = 0.3624; Figure 4c, and IL-10, F(7, 19) = 1.043, p = 0.4351; Figure 4d, relative to sham levels, the increases did not reach statistical significance.
TBI initiated a central and peripheral inflammatory response. Cortical cytokine and chemokine levels were measured by multiplex ELISA following TBI (a-d). Acute increases in proinflammatory (a) IL-6 and (b) CXCL1 were significant in comparison to uninjured sham levels (n = 3–5, mean ± SEM). Serum cytokine and chemokine levels were measured for nine chemokine and cytokine analytes at seven time points following diffuse TBI (e). Proinflammatory CXCL1 and IL-6 were significantly elevated compared to sham during the acute phase following injury (n = 3–5). Although the levels for the remaining molecules changed with respect to sham, the differences were not significant in comparison to sham. Color gradations indicate the protein levels compared to sham, where reds are increased levels and blues are decreased levels.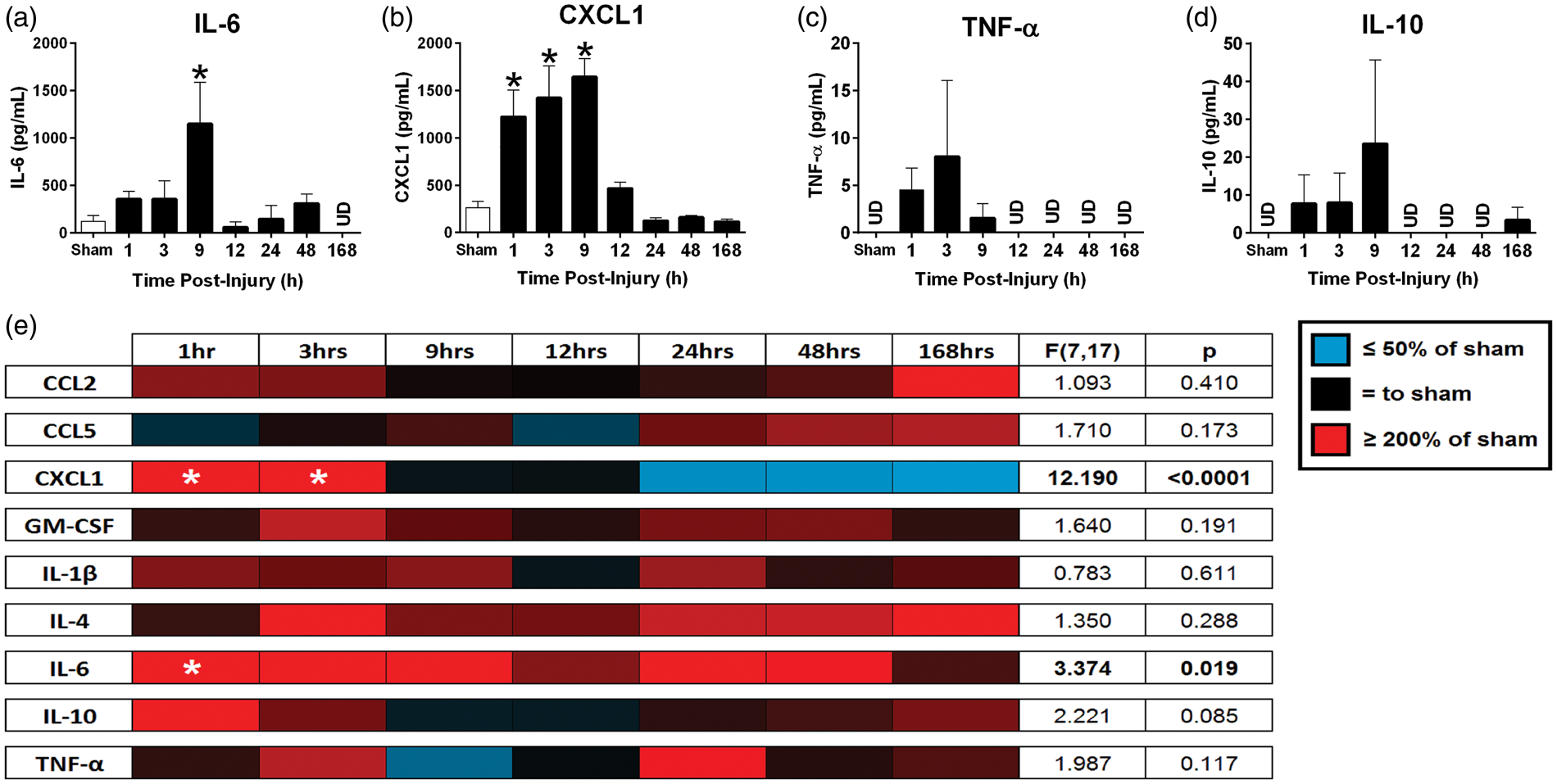
As the BBB is permeable after mFPI, 35 the potential exists for inflammatory molecules produced at the central injury site to act at distal sites in the periphery. To evaluate brain to blood transfer of signal, we repeated and expanded the cytokine array using serum samples. Serum levels of IL-6, F(7, 17) = 3.374, p = 0.019, were significantly different over time postinjury (Figure 4e). Dunnett’s multiple comparison test indicated significant increases in serum IL-6 levels at 1 h postinjury compared to uninjured sham. CXCL1 was significantly different over time postinjury, F(7, 17) = 12.190, p < 0.0001, Figure 4e). IL-4, F(7, 18) = 1.522, p = 0.2225, and IL-10, F(7, 18) = 2.111, p = 0.0956, levels remained increased in serum from brain-injured animals compared to uninjured shams up to one week postinjury, however, these increases did not reach statistical significance.
Diffuse TBI dysregulated induced Treg development
Mesenteric lymph node (MLN) T cells localized into the mucosal immune system showed dysregulated Treg differentiation and proliferation in response to ATRA, a Treg stimulating factor, in brain-injured, but not sham mice (Figure 5). As expected, MLN T cells from uninjured shams (top left, Figure 5a) and brain-injured (bottom left, Figure 5a) mice cultured with dimethyl sulfoxide (DMSO) vehicle showed an activated phenotype (primarily CD25hi/Foxp3lo), with minimal Treg differentiation. MLN T cells from uninjured shams cultured with ATRA, a factor which induces Treg differentiation and mucosal homing, showed increased Treg differentiation (primarily CD25hi/Foxp3hi; top right, Figure 5a). In contrast, MLN T cells from brain-injured mice cultured with ATRA showed minimal T cell activation and less than 1% Treg differentiation (bottom right, Figure 5(a)).
TBI alters regulatory T cell development. CD4+ T cells from mesenteric lymph nodes of brain-injured (n = 4) or sham mice (n = 4) were cultured in media containing DMSO or ATRA, a Treg stimulating factor. (a) T cells were sorted by flow cytometry for traditional markers of T cell activation (CD25) and Treg differentiation (Foxp3). DMSO-treated T cells exhibited a nominal percentage of CD25hi/Foxp3hi Tregs (values displayed in upper right quadrant of each panel). In T cells from sham mice, ATRA stimulation elicited an increase in Treg differentiation that was not observed in brain-injured mice. (b) T cell proliferation was assayed using CFSE assay indicating a reduction in proliferation capacity among T cells from brain-injured mice regardless of media additive (percent of population composed of proliferated cells indicated).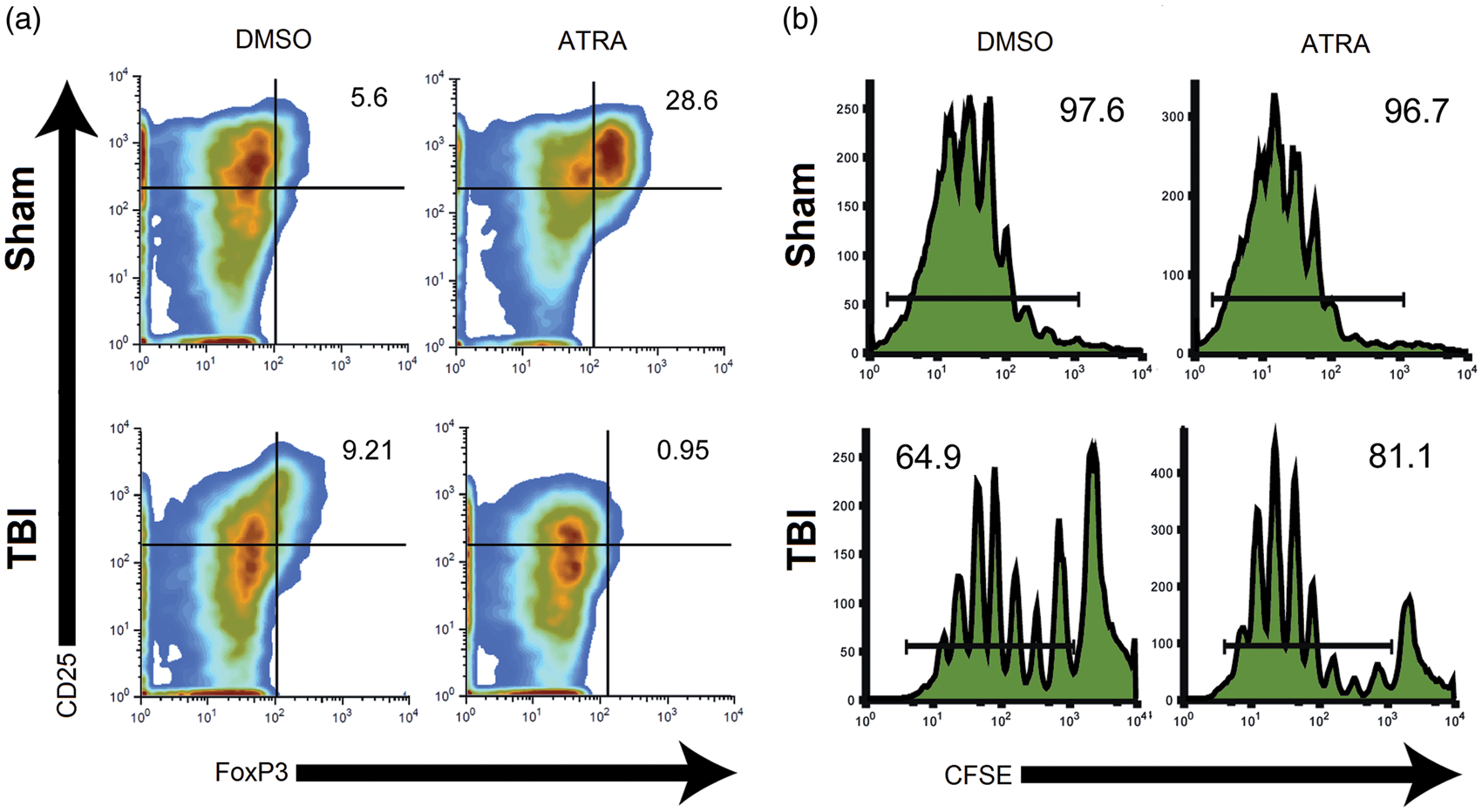
T cells were labeled with CFSE, a fluorescent dye, to ascertain the capacity for cell proliferation. The CFSE proliferation assay indicated lymph cells from uninjured shams demonstrated near maximal proliferation capacity when cultured with either DMSO (97.6% proliferation) or ATRA (96.7% proliferation; top, Figure 5b). However, T cells from brain-injured mice showed a diminished capacity for proliferation when cultured with DMSO (64.9% proliferation) or ATRA (81.1% proliferation; bottom, Figure 5b).
Discussion
TBI may potentiate peripheral inflammatory pain through unregulated systemic inflammation
We show that TBI potentiated carrageenan-induced mechanical hyperalgesia. Our results are the first to advance the idea that TBI-associated increases in inflammation and inflammatory mediators contribute to the potentiation of hyperalgesia following a peripheral immune challenge. 13 TBI activates microglia and astrocytes.36,37 Microglia are resident immune cells of the brain which respond to pathological insult and tissue injury. Following TBI, microglia respond to insult by proliferating and undergoing a phenotypic shift toward “activation.” Activated microglia synthesize and release cytokines into the extracellular environment which have a dual role as inflammation mediators and pronociceptive mediators. 9 Similarly, following a change in the environment such as mechanical injury associated with TBI, astrocytes shift to a reactive phenotype and undergo hypertrophy. In the current study, we demonstrate brain injury-induced activation of both cortical microglia and astrocytes at seven days postinjury. We have previously shown cortical glia activation in the mouse as early as 6 h postinjury 33 supporting rapid glia activation that persists through seven days postinjury. While this is not a novel finding, it is an important confirmation of central inflammation at seven days postinjury in the same animals presenting with hyperalgesia following a peripheral immune challenge.
In the context of experimental and clinical brain injury, glial activation serves as a hallmark of the pathology. Glial activation may lead the injury and repair processes or simply serve as a marker of pathology. In fact, the peripheral increase in circulating GFAP is a leading fluid-based biomarker for detecting brain injury, 38 further demonstrating glial activation and transfer of molecules between central and peripheral compartments. Days following TBI, immune cross talk can likely augment central and peripheral inflammation, which can further contribute to increased sensitivity to secondary challenges including subsequent injuries, stressors, and infections. 39 Following diffuse TBI in mice, microglia are primed, to a heightened state of activation, and this brain-injury-induced inflammatory response has been shown to result in an augmented response to a subsequent peripheral immune challenge one month postinjury. 39 These mice were subjected to mFPI and lipopolysaccharide (LPS) injection (30 days post-FPI), which demonstrated increased depression-like behavior in comparison to mice subjected to either FPI or LPS alone. 39 Similarly, in the current study, we show mFPI in mice activated microglia which may prime them for a secondary challenge (carrageenan injection).
Leakage of cytokines and chemokines from the CNS to the periphery may contribute to TBI potentiation of peripheral inflammatory pain
Under normal conditions, the pain pathway is activated to induce an active and protective response to help prevent further tissue damage. 8 However, when injury and inflammation are prolonged, the ongoing excitation of primary nociceptive neurons can lead to chronic and maladaptive pain. 11 Previous studies have shown in vivo injections of IL-1β, IL-6, or TNF-α induce an active inflammatory event similar to a carrageenan challenge and result in hyperalgesia.40–42 More direct, intraplantar injection of IL-6 into naïve animals has been shown to induce mechanical hyperalgesia. 43
Following brain injury, an inflammatory response is generated by activated glia, which leads to a proinflammatory cytokine response. The resultant inflammatory response is further amplified by migrating blood cells. 13 Proinflammatory cytokines excite nociceptive fibers and increase neural sensitivity.11,17,44–46 Thus, cytokines are not merely inflammatory mediators, but are also neuromodulators, by having direct excitatory actions on peripheral nociceptive neurons. In the current study, diffuse TBI increased cortical and serum levels of cytokine IL-6 and chemokine CXCL1. Based on these observations, we suggest that diffuse TBI induced a CNS inflammatory response, which transitioned rapidly into secreted inflammatory cytokines found in serum, which could potentiate inflammatory pain (Figure 1). Thus, TBI may lead to an acute increase in proinflammatory molecules in the brain that then leak to the periphery, thereby increasing levels in the serum. The ensuing cytokine signaling then potentiates hyperalgesia, as observed.
It is important to note that in the absence of the peripheral inflammation induced by the carrageenan injection, we observed no change in sensory thresholds in mFPI-injured mice compared with uninjured shams at seven days postinjury. This differs from a recent study where TBI induced by lateral fluid percussion (later fluid percussion injury [lFPI]) led to sustained nociceptive sensitization for four days postinjury, without provocation. 47 lFPI is characterized by both focal and diffuse injury in which mechanical damage results in cell death and the development of a cavitation.48,49 It is possible the disruption of the dura and formation of a cavitation extensively disrupts the BBB and initiates a more profound peripheral inflammatory response in the first week postinjury compared with mFPI in the mouse. 48 Further investigation is necessary to compare peripheral inflammation following experimental mFPI and lFPI. In the present case of mFPI, a widespread diffuse brain injury may prime the immune response; whereby, a second inflammatory challenge may be needed to potentiate peripheral inflammatory pain. In contrast, focal injury may substantially increase peripheral inflammation, thus potentiating peripheral inflammatory pain in the absence of a second inflammatory challenge.
Suppressed proliferation and differentiation of Tregs may contribute to pain and dysregulated inflammation following TBI
Our results indicate that MLNs from brain-injured mice cultured with the Treg stimulating factor ATRA failed to differentiate into Tregs. Although ATRA enhances the TGFβ-dependent anti-inflammatory Treg cell differentiation and stability,50,51 addition of ATRA in a proinflammatory context (e.g. IL-15-enriched intestinal mucosa) may promote the inflammatory response instead. 52 Further studies will be necessary to elucidate whether TBI may favor a particular immunological environment in MLNs. Treg differentiation is vital for controlling adaptive and innate immune responses and may also attenuate neuropathic pain. Previous studies have shown neuropathic pain induced by experimental autoimmune neuritis can be attenuated by increasing Tregs, 12 thus the inability of T cells to differentiate into Tregs could contribute to hyperalgesia following experimental TBI.
Our results also indicated that CD4+ MLN T cells from brain-injured mice had suppressed proliferation compared to MLN T cells from sham mice. It has been shown CD4+ T cells from postseptic mice isolated and assayed for ex vivo proliferation following acute inflammation demonstrated deficiencies in proliferative capacity.53,54 Similarly, thermal injury resulted in impaired T cell proliferation in mice. 55 Clinically, 18–72 h after severe TBI, the number of circulating T cells, T-helper cells, and Tregs were reduced.56,57 Hence, an inflammatory challenge, or injury, could lead to cytokine dysregulation, which impairs lymphocyte proliferation, suppresses T cell activation, and alters cell-mediated responses.53–55 In the current study, we show increased cytokine production following diffuse TBI. The resulting systemic proinflammatory environment post-TBI may prime immune cells for a rapid and efficient response against any challenge that may momentarily put a vulnerable, vital organ such as the brain at high risk. Under these conditions, disruption of the Treg cell homeostasis post-TBI, as seen in this study, may support a strong immune response against potential harmful agents. However, it may also prevent the protective role of Tregs in controlling the exacerbated inflammatory reaction to an otherwise weak secondary challenge. Further studies are necessary to elucidate the contribution of brain injury on T cell activation and fate decisions.
Conclusion
In summary, diffuse brain injury led to hyperalgesia following a carrageenan injection in the paw, potentially caused by increased glial activation, central and peripheral increases of inflammatory mediators, and dysregulation of Treg induction. A potential limitation of the current study is the number of animals used in each group, thus, future experiments are needed to elucidate directly the role of Tregs in chronic pain after TBI. Further experiments investigating mechanical pain thresholds post-TBI in other inflammatory pain models such as formalin are of interest but beyond the scope of this experiment. Future experiments are needed to comprehensively investigate inflammatory pain models following experimental diffuse brain injury. Targeting posttraumatic inflammation may reduce secondary injury and improve patient outcome, thus investigation into Treg therapy may be a potential therapeutic target for diffuse brain injury. Furthermore, research is needed to reveal the impact of TBI-induced cytokines and chemokines in the brain on primary afferent nociceptors. Although current studies focus on how glial activation in the spinal cord modulate pain pathways, a lack of studies evaluating how glial activation following brain injury contributes to pain warrants further investigation.
Footnotes
Acknowledgments
The authors thank Greg Bauman and Jennifer Strange for technical assistance.
Authors’ contributions
RKR and JLH carried out the mouse surgeries and injuries, tissue collection, immunohistochemical staining, and data analyses. GIE, RKR, BKT, JL, and FM made substantial contributions to the conception and study design and GIE and FM contributed to peripheral immune data collection and analyses. GIE and GFC carried out the von Frey filament test. ADB and LVE carried out the cytokine measurements in the cortex and serum and analyzed these data. BKT made substantial contributions to the interpretation of the data and made critical revisions to the intellectual content of the manuscript. RKR, GIE, and JL drafted the manuscript and all authors provided revisions and approved the final version. RKR and GIE contributed equally to the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by NIH grants K02 DA19656 and R01 DA037621 (BKT) and R01 NS065052 (JL). Dr. Rowe is funded by a Bisgrove Scholar Award from Science Foundation Arizona. Mr. Harrison was partially supported by the Diane and Bruce Halle Foundation and NIH F31 NS09092. Flow cytometry was carried out at the University of Kentucky Flow Cytometry and Cell Sorting Core Facility, which is supported in part by the Office of the Vice President for Research, the Markey Cancer Center and a grant from the NIH Shared Instrument Program (S10 RR026827).