
Abstract
Background
Chronic pain is often accompanied by short-term memory deficit and depression. Currently, it is believed that short-term memory deficit and depression are consequences of chronic pain. Here, we test the hypothesis that the symptoms might be caused by overproduction of interleukin-1beta (IL-1β) in the injured nerve independent of neuropathic pain following spared nerve injury in rats and mice.
Results
Mechanical allodynia, a behavioral sign of neuropathic pain, was not correlated with short-term memory deficit and depressive behavior in spared nerve injury rats. Spared nerve injury upregulated IL-1β in the injured sciatic nerve, plasma, and the regions in central nervous system closely associated with pain, memory and emotion, including spinal dorsal horn, hippocampus, prefrontal cortex, nucleus accumbens, and amygdala. Importantly, the spared nerve injury-induced memory deficits, depressive, and pain behaviors were substantially prevented by peri-sciatic administration of IL-1β neutralizing antibody in rats or deletion of IL-1 receptor type 1 in mice. Furthermore, the behavioral abnormalities induced by spared nerve injury were mimicked in naïve rats by repetitive intravenous injection of re combinant rat IL-1β (rrIL-1β) at a pathological concentration as determined from spared nerve injury rats. In addition, microglia were activated by both spared nerve injury and intravenous injection of rrIL-1β and the effect of spared nerve injury was substantially reversed by peri-sciatic administration of anti-IL-1β.
Conclusions
Neuropathic pain was not necessary for the development of cognitive and emotional disorders, while the overproduction of IL-1β in the injured sciatic nerve following peripheral nerve injury may be a common mechanism underlying the generation of neuropathic pain, memory deficit, and depression.
Keywords
Background
Chronic pain, short-term memory deficit (STMD), and depression are debilitating neurological diseases and are resistant to currently available therapeutic agents. Chronic pain is often accompanied by STMD1,2 and/or depression.3–6 Despite extensive studies for decades, the mechanisms underlying the comorbidity are still poorly understood.
It is generally believed that chronic pain may impair memory by attracting the limited attention.7,8 However, there are also clinical studies showing that pain does not affect memory in healthy individuals 9 and that relief of pain with opioids cannot improve the memory deficit in chronic pain patients. 10 Likewise, depressive behavior is considered to be a consequence of chronic pain.11,12 On the other hand, it is also proposed that chronic pain and depression share common pathological mechanisms but are independent diseases without causal interaction.13,14 The hypothesis, however, has been largely ignored and never been tested experimentally. Understanding the causal relationship between chronic pain and the memory/emotional disorders is important from both a scientific and clinical perspective. To verify hypothesis, the common pathological mechanisms should be identified.
Interleukin-1 beta (IL-1β), a key cytokine released from glial cells, is critically involved in the pathogenesis of chronic pain, 15 memory deficit, 16 and depression17–19 via activation of IL-1 receptor type 1 (IL-1R1).20,21 For example, intrathecal22,23 or peri-sciatic application 24 of IL-1β is sufficient to induce persistent behavioral signs of neuropathic pain, a common form of chronic pain. IL-1β is remarkably elevated in the hippocampus of human patients with Alzheimer’s disease 25 and IL-1β at pathological concentration inhibits hippocampal long-term potentiation (LTP) and impairs learning and memory.26,27 IL-1β is also elevated in the plasma of human patients with major and postviral depression. 28 Genetic deletion of IL-1R1 prevents depression-like symptoms induced by chronic mild stress in mice. 29 The data from different research fields suggest that overproduction of IL-1β is probably a common pathological mechanism underlying neuropathic pain, memory, and emotional deficits.
Impairment of hippocampus and prefrontal cortex (PFC) is closely associated with both STMD2,30,31 and depression 32 while the dysfunction of amygdala (AMG) 33 and nucleus accumbens (NAcc) 34 are critically involved in depression. Under neuropathic pain conditions, IL-1β mRNA 6 and protein35,36 in spinal cord is upregulated following spared nerve injury (SNI). The change in IL-1β expression in the hippocampus remains controversial, with some studies showing that IL-1β mRNA is upregulated in contralateral hippocampus after SNI but not in chronic constriction injury (CCI) 37 or spinal nerve ligation models, 38 while another study showed bilateral upregulation of IL-1β in the hippocampus in both SNI and CCI models of neuropathic pain. 39 The mRNA expression of IL-1β was also found to be upregulated in the contralateral PFC following SNI.5,40 Little is known whether IL-1β is changed in nucleus accumbens (NAcc) and in AMG in neuropathic pain condition. Thus far, in neuropathic pain model, the temporal and spatial expression pattern of IL-1β at protein level in nervous system has not been investigated systematically and the causal relationship between overproduction of IL-1β and the memory/emotional disorders has not been determined.
To verify the hypothesis that upregulation of IL-1β is a common cause for chronic pain, STMD, and depressive behavior in neuropathic conditions, in the present work we tried to answer following key questions with use of SNI model: Is neuropathic pain associated with STMD and depression? Is overproduction of IL-1β induced by peripheral nerve injury was necessary and sufficient for generation of neuropathic pain, STMD, and depression?
Methods
Animals
Adult male Sprague-Dawley (SD) rats (200–250 g), adult male C57BL/6 mice (23–27 g) were obtained from the Institute of Experimental Animal of Sun Yat-Sen University. IL-1R1 KO mice were purchased from the Jackson laboratory. Adult male IL-1R1 mice (23–27 g) were used for our experiments. The animals were housed individually with free access to food and water ad libitum in a room maintained on a 12/12 h light/dark cycle. The temperature and humidity were kept at 24 ± 1℃ and 50%–60%, respectively. All experimental procedures were approved by the Local Animal Care Committee of Sun Yat-Sen University.
SNI model and behavioral tests
SNI was performed as described by Decosterd and Woolf. 41 Under chloral hydrate anesthesia (0.4 g/kg, i.p.), the three peripheral branches of the sciatic nerve of the left hind limb were exposed. The common peroneal and the tibial nerves were ligated and cut (2 mm sections removed), but the sural nerve was kept intact. At last, the surgical incision was sutured in two layers. For the sham procedure, the three peripheral branches of the sciatic nerve were exposed without any nerve damage.
The paw withdrawal thresholds were measured before and 3, 6, 9, 12 days after SNI and short-term memory recognition index in novel object recognition test and floating time in forced swimming test were assessed before and 12 to 15 days after SNI. All behavioral assays were performed by experimenters who were blind to the experiment designs.
Intravenous injection of rrIL-1β and peri-sciatic application of IL-1β antibody
Recombinant rat IL-1β (rrIL-1β, R&D Systems) was stored as a stock solution (100 µg/ml) at –80℃ and diluted to 100 ng/ml in 0.1% BSA in saline before administration. Tail-intravenous injection of rrIL-1β (100 ng/ml) or vehicle (0.1% BSA in saline) 150 μl in volume was performed three times per day for successive seven days in naïve rats.
To administrate anti-IL-1β (Abcam) or IgG (R&D System) locally to rat sciatic nerve, a permanent indwelling peri-sciatic catheter system was installed, as described before.24 Briefly, sterile gel foam was cut into 15 mm (L) × 5 mm (W) × 10 mm (H) strips aseptically. One end was intersected (3 mm W) to a depth of 1 cm to allow a 5-cm sterile polyethylene (PE-10) tube to be stitched inside. Then the assembly was implanted around the left injured sciatic nerve at mid-thigh level. The operation was performed under anesthesia with chloral hydrate (0.4 g/kg, i.p.) and special care was taken to prevent infection and minimize inflammation. Anti-IL-1β or IgG (80 ng in 200 μl) was injected through the PE-10 tube three times a day for seven days.
Western blotting
Under chloral hydrate anesthesia (0.4 g/kg, i.p.), the sciatic nerve, tissues of lumbar SDH in injured side were collected in SNI and sham-operated animals. Brains removed from decapitated animals were sectioned at 500 µm in a cryostat to collect NAcc, PFC, hippocampus, and AMG in the contralateral side based on the rat and mouse brain atlas. In the rats with intravenous injection of IL-1β, the nerve tissues from both sides were collected. The tissues were homogenized and sonicated on ice in lysis buffer with protease inhibitor cocktail (Roche Molecular Biochemicals) and phosphatase inhibitor, followed by centrifugation at 13,800 g for 20 min at 4℃ to obtain supernatant containing protein. Equal concentration of protein from different supernatant was loaded and separated by SDS-PAGE. After the transfer to a PVDF membrane (Bio-Rad), blots were blocked and incubated at 4℃ overnight with primary antibodies including rabbit anti-IL-1β (Abcam, ab2105, 1:1000) and mouse anti-β-actin (Cell Signalling Technology, 1:1000). After incubated with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG (secondary antibody, 1:8000 or 1:10000, Jackson Immune laboratory) for 1 h at 25℃ and washed, the immune complex on the membrane was detected by enhanced chemiluminescent (Bio-Rad) and exposed by the Tanon-5200 Chemiluminescent Imaging System (Tanon Science and Technology). The density of specific bands representing protein content was quantified with an image analysis program (Image Pro Plus).
IL-1β bioassay
Under urethane (1.4 g/kg, i.p.), rat blood was sampled by eyeball extirpating at different time points after SNI (5 h to 14 days, see Figure 2(i)). After 30 min standing at room temperature, the upper layer of the serum was collected and was centrifuged for 12 min (3,000 rpm, 4℃). The sciatic nerve from the ipsilateral side was harvested in the same cohort of rats and was homogenized in ice-cold PBS followed by centrifugation at 4℃ for 20 min (12,000 rpm, 4℃). The supernatants of samples were used to measure the concentrations of IL-1β immediately by using anti-rat IL-1β ELISA Kits (R&D system), according to the manufacturer’s specification.
Immunohistochemistry
Animals were anesthetized with chloral hydrate anesthesia (0.4 g/kg, i.p.) and perfused transcardially with 100 ml of PBS followed by 200 ml of ice cold 4% paraformaldehyde (PFA) in PBS containing 1.5% picric acid. The spinal cord and brain were isolated and post-fixed with the same 4% PFA for 4 to 6 h at 4℃. The samples were then transferred to 30% sucrose in PBS overnight. Cryosections 20 µm in thickness were made using a cryostat (Leica) and laid on a gelatin-coated glass slide. The sections were blocked with 5% goat serum and 0.3% Triton X-100 (Sigma) in TBS buffer for 60 min and then incubated overnight at 4℃ with primary antibody for rabbit-anti-Iba1 (1:500, Abcam). The sections were then incubated for 1 h with FITC-conjugated secondary antibodies at room temperature (1:500, Jackson ImmunoResearch). The fluorescent images were obtained with a Leica DFC350 FX (Leica Camera AG, Solms, Germany) fluorescence microscope and images were captured with a CCD spot camera.
Statistical Analysis
All data were presented as mean ± SEM. The results of the novel object recognition test, forced swim test, Western blot, and IL-1β bioassay were compared using one-way analysis of variance (ANOVA) followed by Turkey analysis. For paw withdrawal threshold, the data were analyzed with repeated measures two-way ANOVA, and post hoc test were used for detailed statistical analysis when appropriate. The correlation coefficients were calculated to determine the relationship between paw withdrawal threshold and recognition index or floating time. All experiments were repeated three to four times. Data were analyzed with SPSS 16.0. P < .05 was considered to be statistically significant.
Results
The mechanical allodynia is correlated with neither STMD nor depressive behavior SNI rats
Compared with sham group, the paw withdrawal thresholds in bilateral sides and STM recognition index decreased and floating time increased significantly in SNI rats (Figure 1(a)–(c)), indicating that mechanical allodynia (a behavioral sign of neuropathic pain), STMD, and depressive behavior were developed in SNI rats. The results are in consistence with previous studies.2,6,41 To investigate whether neuropathic pain is associated with STMD or with depressive behavior, correlation analysis was performed in the 44 SNI rats. As shown in Figure 1(d) and (e), paw withdrawal thresholds were correlated with neither STM recognition index nor floating time. The data do not support the point of review that chronic pain leads to memory deficit7,8 and depressive behavior.11,12
Mechanical allodynia, memory deficit, and depressive behavior induced by SNI in rats. (a) The time courses of paw withdrawal thresholds on ipsilateral (Ipsi) and contralateral (Contra) sides in SNI (n = 44) and sham-operate (n = 38) rats are shown. Operations were performed on day 0. The short-term memory index assessed with novel object recognition test (b) and the floating time with the forced swim test (c) in SNI group and in sham group were measured before and 12 to 15 days after SNI. **P < .01; ***P < .001 vs. sham group; ##P < .01 vs. SNI group before operation. (d, e) Paw withdrawal thresholds in ipsilateral side measured at 12 days after SNI were correlated with neither short-term memory index nor the floating time in the 44 SNI rats. IL-1β is increased in the injured sciatic nerve, plasma, spinal dorsal horn, hippocampus, prefrontal cortex, nucleus accumbens, and amygdala following SNI. (a–f) Western blots show IL-1β expression in different nerve tissues as indicated in sham and 1 day, 3 days, and 7 days after SNI in rats (n = 5–6 in each group). (g) The specificity of anti-IL-1β used in the present study was identified by pre-incubation with rrIL-1β. The pre-incubated antibody detects very weak signal in blot with proteins from injured sciatic nerve. (h and i) The concentrations of IL-1β in the injured sciatic nerve tissue and plasma at different time points following SNI detected by ELISA (n = 6–7 in each group). **P < .01; ***P < .001 vs. sham group. SNI: spared nerve injury; SDH: spinal dorsal horn; NAcc: nucleus accumbens; AMG: amygdala.

Temporal and spatial expression profiles of IL-1β in the injured sciatic nerve, plasma, and central nervous system (CNS) following SNI
Several lines of evidence from different research fields have demonstrated that the overproduction of IL-1β in different brain regions plays a key role in neuropathic pain, 15 memory deficit, 16 and depression.17–19 To test whether upregulation of IL-1β is a common cause for chronic pain, STMD, and depressive behavior in neuropathic conditions, we measured the IL-1β protein in plasma, the ipsilateral sciatic nerve, spinal dorsal horn (SDH), contralateral hippocampus, PFC, NAcc, and AMG at different time points following SNI using ELISA and Western blots. Compared to sham group, the level of IL-1β in the injured sciatic nerve was around eight folds higher on day 1 and three folds higher on day 3 to 7 in SNI rats (Figure 2(a)). While, significant increase in IL-1β protein was detected in SDH, PFC, and AMG starting on day 3 (Figure 2(b), (d), and (f)) and in hippocampus and NAcc starting on day 7 after SNI (Figure 2(c) and (e)). The specificity of anti-IL-1β used in the present study was identified by pre-incubation with rrIL-1β (Figure 2(g)).
ELISA experiment confirmed the upregulation of IL-1β protein in the injured sciatic nerve. The concentration of IL-1β in the injured sciatic nerve tissue increased to around three folds at 5 h and to eight folds at 12 h following SNI and remained at the peak level at 24 h (Figure 2(h), P < .001 vs. sham rats). The expression of IL-1β protein in the injured sciatic nerve decreased gradually at 3, 7, and 14 days after SNI but was still much higher than that in sham group (P < .01). We also measured the concentration of IL-1β in plasma in the same cohort of rats. A significant increase was detected at 5 h (Figure 2(i), P < .01vs. sham rats), reaching to peak on day 3 (P < .001), remaining at high levels on day 7 and day 14 after SNI (P < .001). These results demonstrate that SNI upregulates IL-1β first in the injured sciatic nerve (5 h), then in plasma (1 day) and finally in the different brain regions (≥3 days), suggesting the injured sciatic nerve is the initiation site for IL-1β upregulation.
Anti-IL-1β rescues behavioral deficits induced by SNI
To determine the causal relationship between the upregulation of IL-1β and the behavioral abnormalities in SNI animals, we performed two series of experiments using IL-1β neutralizing antibody or genetic deletion of IL-1R1. We first tested whether the increased IL-1β in injured sciatic nerve serves as a trigger for SNI-induced behavioral changes by local application of IL-1β neutralizing antibody at the injured nerve. Anti-IL-1β or IgG (80 ng in 200 μl) was administrated 30 min before and three times daily for seven successive days after SNI via a permanent indwelling peri-sciatic catheter system. The mechanical allodynia was then assessed 3 to 12 days after onset of injection and other behavioral tests were performed at 7 to 10 days after last application of the antibody or IgG. We found that the anti-IL-1β did not affect the paw withdrawal threshold in sham-operated rats (Figure 3(a) and (b), P > .05, Con + Sham vs. I-Anti + Sham group), but significantly increased the paw withdrawal threshold in both ipsilateral and contralateral sides in SNI rats, when applied to operated side. Furthermore, we found that the local application of anti-IL-1β at injured side also prevented the decrease in recognition index and increase in floating time induced by SNI (Figure 3(c) and (d), P < .001, I-Anti + SNI vs. I-IgG + SNI groups) but did not affect the parameters in sham rats (Figure 3(c) and (d), P > .05, Con + Sham vs. I-Anti + Sham group). To verify that primary effect of anti-IL-1β on behavioral abnormities is at the injured nerve, the same dose of antibody was applied into the contralateral sciatic nerve with the same method in another seven SNI rats, and no effect on the behavioral deficits induced by SNI was detected (Figure 3(a)–(d), P > .05, C-Anti + SNI vs. I-IgG + SNI groups).
Peri-sciatic application of IL-1β neutralizing antibody at injured sciatic nerve prevents the abnormal behaviors induced by SNI in rats. Paw withdrawal threshold (a, b), recognition index for STM (c), and floating time (d) in different groups in rats are shown (n = 7–12/group). Anti: Anti-IL-1β; I: ipsilateral; C: contralateral; no antibody and IgG were applied in Con + sham group; *P < .05; **P < .01 ***P < .001 vs. Con + sham group; #P < .05; ##P < .01; ###P < .001 vs. I-IgG + SNI group. SNI: spared nerve injury.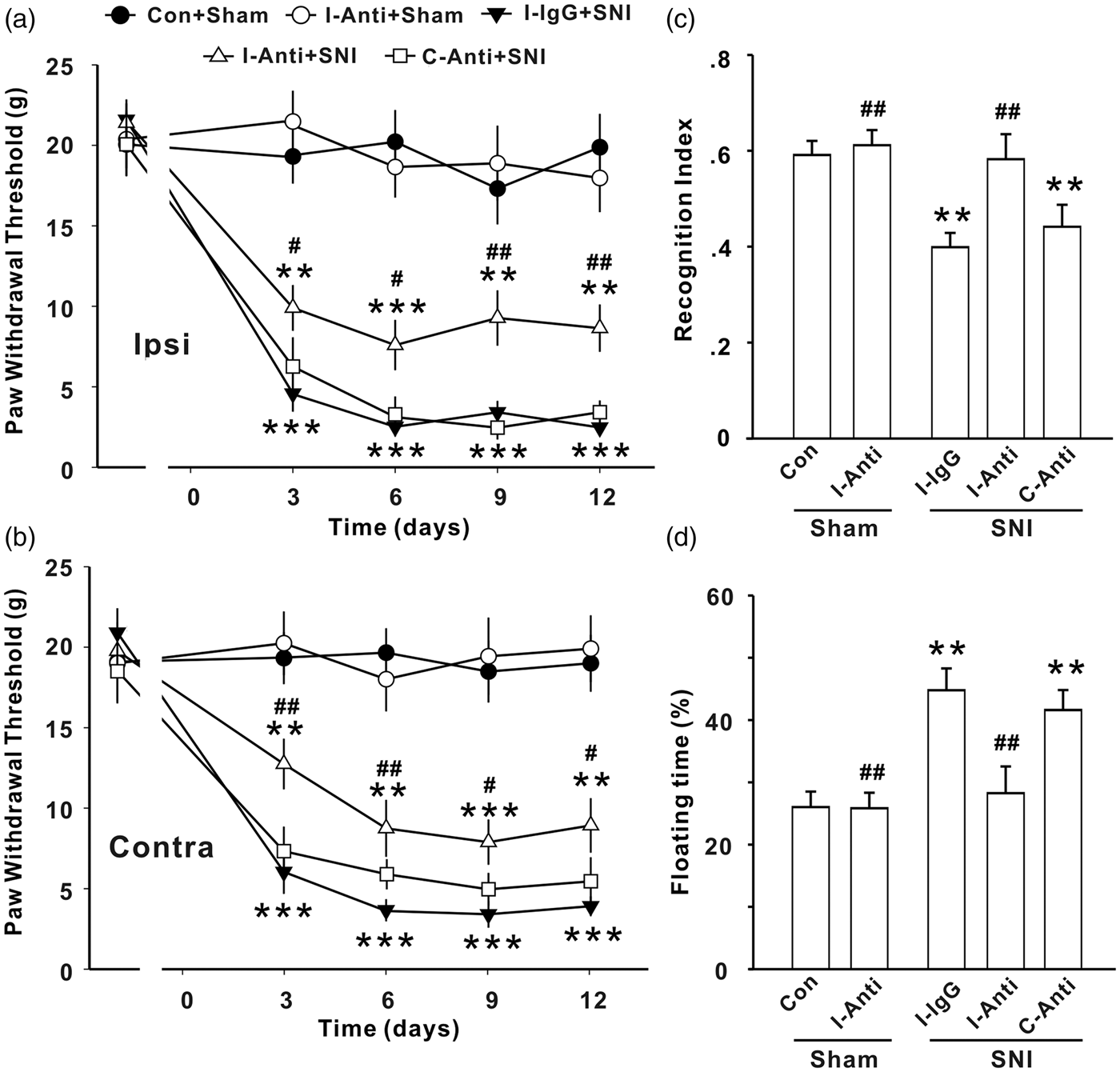
To test whether the increased IL-1β in the injured sciatic nerve may also trigger the upregulation of the cytokine in CNS, we assessed IL-1β levels in SDH, hippocampus, PFC, NAcc, and AMG with Western blots following peri-sciatic application of anti-IL-1β at injured sciatic nerve. Indeed, we found that IL-1β levels in all brain regions in the SNI rats treated with the anti-IL-1β were lower than those in IgG-treated groups (Figure 4(a) and (b), P < .001). The data suggest that the IL-1β upregulation in CNS started in the injured sciatic nerve may contribute to the development of neuropathic pain, STMD, and depressive behavior following peripheral nerve injury.
Peri-sciatic application of anti-IL-1β at injured sciatic nerve prevents the overexpression of IL-1β induced by SNI in rats. (a, b) The Western blots show the expression of IL-1β in SDH, hippocampus, PFC, NAcc, and AMG in the different treatment groups as indicated (n = 5–6 in each group). Anti: Anti-IL-1β; *P < .05; **P < .01 ***P < .001 vs. sham group; #P < .05; ##P < .01; ###P < .001 vs. IgG + SNI group. SNI: spared nerve injury; SDH: spinal dorsal horn; NAcc: nucleus accumbens; AMG: amygdala.
IL-1R1 deficiency reverses SNI-induced neuropathic pain, memory deficits, and depressive behaviors
To confirm that IL-1β signaling is indeed involved in the SNI-induced behavioral and immune abnormalities, we next performed the experiments with IL-1R1 knockout (IL-1R1 KO) mice. Similar to the results in rats, SNI also caused a significant decrease in paw withdrawal threshold bilaterally in wild-type (WT) mice (P < .001). The thresholds in KO SNI mice were significantly higher than those in WT SNI mice (Figure 5(a) and (b), P < .05) but were still lower compared to naïve WT mice (P < .05), indicating that deletion of IL-1R1 also partially prevented SNI-induced mechanical allodynia. In addition, the reduction in STM recognition index and the increase in floating time induced by SNI were completely prevented in IL-1R1 KO mice two weeks after SNI (Figure 5(c) and (d), P > .05). No difference in parameters between KO mice and WT mice was detected in sham control conditions (Figure 5(c) and (d), P > .05).
Genetic deletion of IL-1R1 prevents both abnormal behaviors and the overexpression of IL-1β induced by SNI in mice. (a–d) Paw withdrawal threshold, recognition index for STM, and floating time in wild type (WT) mice with or without SNI and IL-1R1 knockout (KO) mice with SNI or without SNI are shown (n = 7–12/group). (e) The Western blots show the expression of IL-1β in SDH, hippocampus, PFC, NAcc, and AMG in the different groups as indicated (n = 5–6 in each group). *P < .05; **P < .01; ***P < .001 vs. WT naïve mice; #P < .05; ##P < .01; ###P < .001 vs. WT + SNI mice. SNI: spared nerve injury; SDH: spinal dorsal horn; NAcc: nucleus accumbens; AMG: amygdala.
Compared to sham group mice, SNI mice also showed upregulated IL-1β in SDH, hippocampus, PFC, NAcc, and AMG (P < .001, vs. naïve mice). Interestingly, the SNI-induced IL-1β upregulation in the SDH, hippocampus, PFC, NAcc, and AMG was completely blocked by deletion of IL-1R1 (Figure 5(e), P < .001). There was no difference in IL-1β expression in the naïve hippocampus, PFC, NAcc, and AMG between WT mice and IL-1R1 KO mice (P > .05). Together, the data indicate that IL-1R1 may mediate IL-1β upregulation that is critical for the generation of neuropathic pain, memory deficit, and depressive behavior following SNI.
Intravenous injection of IL-1β at pathological concentration mimics the effects of SNI
To test whether increased IL-1β in plasma observed in SNI rats is sufficient to induce the behavioral abnormalities through the IL-1β upregulation in CNS, Naïve rats were treated with rrIL-1β or vehicle (0.1% bovine serum albumin (BSA) in saline) via tail-intravenous injection three times per day for seven days. As the peak concentration of IL-1β in plasma detected in SNI rat was around 800 pg/ml (Figure 2(i)) and blood volume of adult rat (±200 g) was around 20 ml, 150 μl of rrIL-1β at 100 ng/ml per rat was administrated, to reach around 800 pg/ml in plasma. In 31 rats treated with IL-1β, a significant decrease in paw withdrawal thresholds bilaterally on day 3 after onset of injection (Figure 6(a), P < .01), and no change in paw withdrawal threshold was detected in the rats receiving vehicle injection. The STM recognition index was decreased; the floating time was increased significantly at two weeks after onset of rrIL-1β injection in the same cohort of rats (Figure 6(b) and (c), P < .05 vs. vehicle group). Similar to the results in SNI rats, correlation analysis in rrIL-1β treated rats revealed that paw withdrawal threshold was correlated with neither STM recognition index nor floating time (Figure 6(d) and (e)). Following the behavioral tests, we also assessed the expression of IL-1β in CNS using Western blots. The levels of IL-1β in SDH, hippocampus, PFC, NAcc, and AMG were significantly higher in rrIL-1β-treated rats, compared to vehicle-treated ones (Figure 6(f), P < .01 or P < .001). The behavioral and molecular changes induced by intravenous injection of rrIL-1β were comparable to those induced by SNI in rats (Figures 1 and 2).
Intravenous injection of IL-1β induces mechanical allodynia, memory deficit, and depressive behavior and upregulates IL-1β in CNS in rats. (a) PWTs in bilateral handpaws decreased in the rats receiving tail-intravenous injection of rrIL-1β (100 ng/ml, 150 μl in volume per rat, t.i.d, for 7 days, n = 31) but not in the rats with vehicle injection (n = 30) in the same way. rrIL-1β or vehicle was injected at day 0. (b, c) The recognition index for STM and the floating time measured two weeks after first rrIL-1β or vehicle injection in the same cohort of rats are shown. (d, e) Paw withdrawal thresholds in left side measured at 13 days were correlated with neither short-term memory index nor the floating time in the 31 rats receiving injection of rrIL-1β. (f) The expression of IL-1β in different regions of CNS was increased as assessed after behavioral tests. *P < .05; **P < .01 ***P < .001 vs. vehicle group. SDH: spinal dorsal horn; NAcc: nucleus accumbens; AMG: amygdala.
Microglia are activated by peripheral IL-1β signaling
In response to pathological stimuli, microglia are rapidly activated, undergo morphological and functional alterations, and release pro-inflammatory cytokines, including IL-1β.43,44 However, whether brain microglia are activated by peripheral nerve injury is still debated.
45
To address this question, we first examined microglial activation in different brain regions using Iba1 staining. Compared to the sham group, the expression of Iba1 on day 12 in SNI rats was significantly higher not only in SDH but also in hippocampus CA1, PFC, NAcc, and AMG (Figure 7(a), (d), (e)). In addition, the proportion of hypertrophic Iba1-positive cells with thick and radial processes increased in SNI group. These results showed that CNS microglia were activated by SNI.
The activation of microglia in the spinal dorsal horn or CNS regions is interfered by the change of peripheral IL-1β signaling. (a–c) Representative pictures showing that microglia were activated in different regions of the CNS at 12 days following SNI (a) and was blocked by peri-sciatic application of IL-1β neutralizing antibody (b) (n = 3/group). Iba1 is a microglial marker. (c) Tail-intravenous injection of rrIL-1β but not vehicle induced the activation of microglia. (n = 3–4/group). (d) Bar graph summarizing the number of Iba1 positive cells area in various regions of the CNS in different treatment groups. *P < .05; **P < .01; ***P < .001 vs. sham group. ##P < .01; ###P < .001 vs. IgG + SNI group. SDH: spinal dorsal horn; NAcc: nucleus accumbens; AMG: amygdala.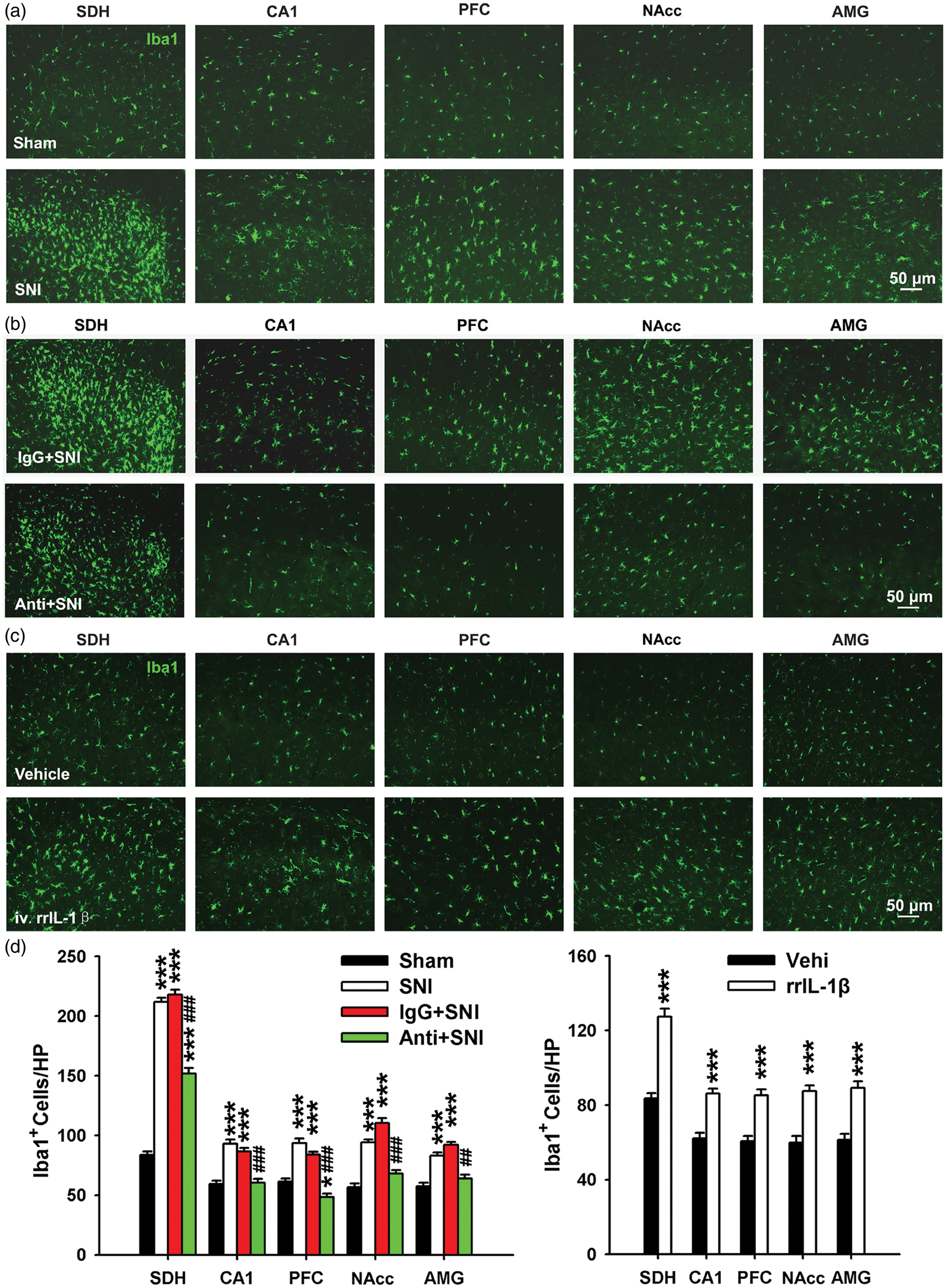
Our data demonstrated that elevated IL-1β expression was first observed in the injured sciatic nerve followed by increased expression in many brain regions after SNI. Since it is known that IL-1β could activate microglia,46,47 we went on to investigate whether upregulated IL-1β in the injured sciatic nerve is necessary to activate CNS microglia. To this end, we tested whether local blockage of IL-1β signaling by peri-sciatic application of anti-IL-1β antibody could prevent microglia activation in the CNS. Continuous application of anti-IL-1β antibody (but not IgG) partially blocked microglia activation in the ipsilateral SDH and completely inhibited microglial activation in many brain areas (Figure 7(b) and (d)). These data indicate that overproduced IL-1β in the injured sciatic nerve may be an initiating factor for CNS microglia activation after SNI.
Finally, we tested whether increased IL-1β in plasma is sufficient to induce CNS microglia activation. Compared to vehicle, intravenous injection of rrIL-1β at pathological concentration for 7 continuous days increased the number of microglia in SDH and hippocampus CA1, PFC, NAcc, and AMG on day 12 (Figure 7(c) and (d)). Therefore, intravenous injection of rrIL-1β also mimics the effects of SNI on brain microglia activation. Taken together, these findings further confirmed that the overproduced IL-1β in injured sciatic nerve may be a common cause for generation of neuropathic pain, STMD, and depression by increased IL-1β in plasma and microglia activation in the brain.
Discussion
The main findings of the present work are following. At first, mechanical allodynia, a behavioral sign of neuropathic pain, was correlated with neither STMD nor depressive behavior in SNI rats; Second, IL-1β, which is known to induce chronic pain, memory and emotion deficits, was upregulated in injured sciatic nerve, plasma, and CNS; Third, the SNI-induced behavioral deficits were prevented by local application of anti-IL-1β at injured sciatic nerve in rats or deletion of IL-1R1 in mice and were mimicked by repetitive intravenous injection of rrIL-1β in rats. The data suggest that neuropathic pain may be dissociated with memory deficit and depression, while the overproduction of IL-1β is likely a common mechanism underlying the abnormal behaviors under neuropathic conditions, although the possibility that disorders may exacerbate each other in the middle and late phase cannot be excluded.
Relationships between neuropathic pain and cognitive/emotional disorders
In the present study, we showed that peripheral nerve injury induced mechanical allodynia, STMD, and depressive behavior in both rats and in mice. In general, our results are consistent with previous animal studies2,48,49 and the clinical reports.10,12 However, we showed that neuropathic pain is not related with memory deficit and depression in the SNI rats and in IL-1β-injected rats. The result is particularly interesting because it suggests that the generation and maintenance of memory deficit and/or depression may be not a direct result of chronic pain in neuropathic condition. This is in line with a recent work showing that the memory deficit and depression evoked by a reversible peripheral neuropathy persists even after resolution of prolonged mechanical allodynia. 49 Furthermore, Zhou et al. 6 have shown that the inhibition of depression-like behavior did not prevent the development of pain, indicating that the dissociation of neuropathic pain and depression after peripheral nerve injury. Taken together, the experimental studies indicate that the symptoms may be independent without causal relationship.
The temporal overproduction of IL-1β from injured sciatic nerve, plasma to CNS after SNI
It has been proposed that IL-1β in plasma is able to directly affect the function of the CNS by crossing the blood-brain barrier (BBB). 50 In addition, IL-1β itself increases BBB permeability in a model of multiple sclerosis. 51 Peripheral nerve injury also increases the permeability of BBB through activation of primary afferent C-fibers 52 and of blood-spinal cord barrier (BSCB) via activation of inflammatory pathway. 53 Here we showed that IL-1β in the injured sciatic nerve was increased at 5 h following SNI, reached to peak at 12 h, and remained at high level at least for two weeks. Likewise, elevation of IL-1β in plasma was detected as early as 5 h, but the peak was reached only at three days after surgery. While a significant increase of IL-1β was detected in the CNS regions at three days or seven days after SNI, much later than in the injured sciatic nerve and plasma (Figure 2). It is plausible that the upregulated IL-1β in the plasma following SNI increased the permeability of BBB and BSCB, thereby plasma may be a source for IL-1β in the CNS. Another source of IL-1β in the CNS may come from the local amplification as it is well established that IL-1β promotes itself production via autocrine and/or paracrine mechanism. 54 Taken together, the elevated IL-1β in the injured sciatic nerve and plasma may lead to the upregualtion of IL-1β in CNS following peripheral nerve injury.
IL-1β overproduction is necessary and sufficient for generation of neuropathic pain, memory deficits, and depression following peripheral nerve injury
It has long been suggested that chronic pain and depression may share common pathological mechanisms.13,14 We believe that overproduction of IL-1β is likely a convergent pathway for the initiation of neuropathic pain, cognitive, and emotional deficits, based on the following evidence. Firstly, the upregulation of IL-1β protein was detected in injured sciatic nerve and in plasma at 5 h and then in SDH, hippocampus, PFC, NAcc, and AMG, which are closely associated with pain, memory, and emotion; Secondly, either local application of IL-1β neutralizing antibody at the injured sciatic nerve (Figures 3 and 4) or deletion of IL-1R1 in mice (Figure 5) substantially blocked mechanical allodynia, STMD, and depression as well as the upregulation of IL-1β in the CNS induced by SNI; Next, intravenous injection of rrIL-1β at a pathological concentration determined in SNI rats mimicked pain hypersensitivities, memory deficits, and depressive behaviors in naïve rats; Finally, we found microglia, which release many proinflammatory cytokines,55 were activated in regions of CNS following SNI, and the effect was substantially attenuated by interruption of IL-1β signaling.
IL-1β mechanism of mirror pain after SNI
Our present work on peripheral IL-1β mechanism may also explain the long-standing puzzle of bilateral pain hypersensitivities following peripheral nerve injury. Original study by Decosterd and Woolf 41 showed that SNI produces mechanical allodynia only in the injured side but not in contralateral side. However, later studies have shown that SNI,56 ligation and transection of sciatic nerve, 57 common perineal nerve ligation, 58 or L5 ventral root transection 59 at one side lead to mechanical allodynia in bilateral hind paws. Similarly, L5 and L6 spinal nerve ligation is primarily reported to induce mechanical allodynia only in injured side in male SD rats, 60 while recent works showed that the same injury model produces bilateral mechanical allodynia in the same gender and strain of rats. 61,62 Our results shed new light on the mechanism of mirror pain and demonstrate that the increased IL-1β produced by local nerve injury could potentially initiate bilateral pain. In line with this notion, intravenous injection of IL-1β was able to induce bilateral pain hypersensitivities. In addition, previous studies showed that intraneural injection of IL-1β at 0.25 pg/ml 23 or peri-sciatic application of IL-1β at 100 pg/ml 24 was sufficient to induce bilateral lasting mechanical allodynia. As the peak concentration of IL-1β reached to 800 pg/ml in plasma in SNI rats (Figure 2(i)), it is not surprising that peripheral nerve injury produces bilateral behavioral signs of neuropathic pain. Taken together, these mirror pain induced by peripheral nerve injury could be due to the local production of IL-1β in sciatic nerve and subsequent propagation into the spinal cord and brain.
Region-dependent synaptic plasticity, a possible mechanism by which SNI or IL-1β induces neuropathic pain, memory deficit, and depressive behaviors
It has been well established that the change in synaptic strength in CNS underlies chronic pain, 63,64 memory formation, 65 and mood depression. 66 Interestingly, accumulating evidence shows that peripheral nerve injury affects synaptic strength in a region-dependent manner. It induces LTP at C-fiber synapses in SDH, 67 contributing to pathological pain, 64 but impairs LTP in hippocampus, leading to working memory and short-term memory deficit. 2 LTP at synapses between hippocampus and PFC is implicated in working memory 68 and emotion. 69 The synaptic connection in this pathway is reduced by SNI. 70 In contrast, SNI potentiates the synaptic transmission between parabrachial nucleus-central nucleus of amygdala 71 and PFC, 69 leading to memory deficit and depressive behaviors. It has been proposed that the reduced excitatory synaptic transmission in both hippocampus- and PFC-NAcc pathways, leading to a dysfunction of corticomesolimbic reward circuitry that underlies many of the symptoms of depression. 72 Consistently, optogenetic activation of the PFC-NAcc pathway inhibits neuropathic pain and the affective symptoms produced by SNI. 73 Several lines of evidence show that proinflammatory cytokines, including IL-1β and TNF-α, regulate synaptic strength also in a region-dependent manner. Both IL-1β 74 and TNF-α 75 are necessary for induction of LTP at C-fiber synapses in SDH. 76,77 While the cytokines at pathological concentration inhibit LTP in hippocampus 78–80 and in frontal cortex. 70 Taken together, peripheral nerve injury and the resultant upregulation of IL-1β may lead to the neuropathic pain, memory deficit, and depression-like behavior via the region-dependent changes in synaptic strength. As neuropathic pain was dissociated with STMD and depression-like behavior in SNI and IL-1β injected rats, we proposed that the changes of synaptic connections in different regions may be variable in a given animals. Further studies are needed for elucidate the mechanisms underlying region-dependent regulation of synaptic strength induced by proinflammatory cytokines.
Conclusions
The upregulation of IL-1β is a common cause for chronic pain, memory deficits, and depressive behavior in neuropathic conditions. Hence, IL-1β may be a target for prevention of neuropathic pain and the accompanied cognitive and emotional disorders.
Footnotes
Authors’ Contributions
WSG, LJW, LJZ, and XGL conceived of the project, designed the experiments. WSG, XW, CLM, and LJZ carried out all experiments. WSG, XW, and MM analyzed the data and prepared the figures. LJZ and XGL supervised the overall experiment. MM, LJW, LJZ, and XGL revised the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (U1201223 and 8137119), Guangdong Province University Outstanding Young Teachers’ Training Program (S2013010011889), and from Natural Science Foundation of Guangdong Province, China (Yq2013008).