
Abstract
Background/Aims:
Existing regulatory and ethical guidance does not address real-life complexities in how clinical trial participants’ level of participation may change. If these complexities are inappropriately managed, there may be negative consequences for trial participants and the integrity of trials they participate in. These concerns have been highlighted over many years, but there remains no single, comprehensive guidance for managing participation changes in ways that address real-life complexities while maximally promoting participant interests and trial integrity. Motivated by the lack of agreed standards, and observed variability in practice, representatives from academic clinical trials units and linked organisations in the United Kingdom initiated the PeRSEVERE project (PRincipleS for handling end-of-participation EVEnts in clinical trials REsearch) to agree on guiding principles and explore how these principles should be implemented.
Methods:
We developed the PeRSEVERE principles through discussion and debate within a large, multidisciplinary collaboration, including research professionals and public contributors. We took an inclusive approach to drafting the principles, incorporating new ideas if they were within project scope. Our draft principles were scrutinised through an international consultation survey which focussed on the principles’ clarity, feasibility, novelty and acceptability. Survey responses were analysed descriptively (for category questions) and using a combination of deductive and inductive analysis (for open questions). We used predefined rules to guide feedback handling. After finalising the principles, we developed accompanying implementation guidance from several sources.
Results:
In total, 280 people from 9 countries took part in the consultation survey. Feedback showed strong support for the principles with 96% of respondents agreeing with the principles’ key messages. Based on our predefined rules, it was not necessary to amend our draft principles, but comments were nonetheless used to enhance the final project outputs. Our 17 finalised principles comprise 7 fundamental, ‘overarching’ principles, 6 about trial design and setup, 2 covering data collection and monitoring, and 2 on trial analysis and reporting.
Conclusion:
We devised a comprehensive set of guiding principles, with detailed practical recommendations, to aid the management of clinical trial participation changes, building on existing ethical and regulatory texts. Our outputs reflect the contributions of a substantial number of individuals, including public contributors and research professionals with various specialisms. This lends weight to our recommendations, which have implications for everyone who designs, funds, conducts, oversees or participates in trials. We suggest our principles could lead to improved standards in clinical trials and better experiences for participants. We encourage others to build on our work to explore the application of these ideas in other settings and to generate empirical evidence to support best practice in this area.
Introduction
It is well established in modern research ethics that clinical trial participants must retain control over their ongoing participation. This is mainly addressed through the ‘right to withdraw consent’ in relevant laws, policies and guidelines, which standardly says that participants may stop taking part at any time, without explanation or reprisal.1–5 Establishment of this right was essential following previous unethical practices and protects participants’ autonomy.6,7
However, the right does not address real-life complexities in the nature and extent of different ‘participation changes’ (used henceforth as a catch-all for any ways that participation can stop, reduce or change). The term ‘withdrawal’ itself, as used in the above-referenced sources, might imply that research participation has two states: continuation or discontinuation. In practice, many clinical trials comprise several participation elements, such as receiving intervention, completing questionnaires, 8 attending follow-up visits or allowing data collection from routine clinic visits or central databases. 9 It is often possible for participants to stop some elements while continuing others. For example, participants may no longer feel able to attend trial-specific clinic visits but may be happy to continue follow-up via another method (e.g. via routine healthcare appointments only). Existing policies also do not explain what should happen when contact is lost between researchers and participants, or when caregivers or those with a duty of care decide it is in participants’ interests for some aspects of participation to stop (e.g. receipt of trial intervention).
These limitations lead to potential challenges in practice. For example, they might lead to the assumption that any expressed participant wish to ‘withdraw’ from a trial must result in all aspects of their participation stopping. If this assumption leads to the loss of potentially available trial outcome data, it may unnecessarily impair trial integrity,10–12 devalue individual participants’ contributions (and those of other participants in the same trial) and could be considered research waste. 13 It may also be unfair to participants who would be happy to continue participating in a more limited fashion or want to stay in touch and find out the results. Alternatively, if participants have the impression that they may only continue or stop all aspects of the trial, some may feel pressure to continue participating to avoid ‘letting the trial down’. 14
This basic ‘right to withdraw’ has remained largely unchanged since the mid-20th century, despite important changes in the relationship between society and research during this time. Clinical research is now more commonly done collaboratively with patients and the public, 15 improving the chances of producing ‘fruitful results for the good of society’. 16 It is also accepted that research participation can benefit individuals, in a broad sense.17–20 By extension, if a participant is struggling to continue with all trial activities, continuing with reduced participation, instead of stopping all trial-associated activity and contact, could also benefit them. Failure to offer participants such reduced options when they should be possible may contradict the idea of participants having the right to withdraw consent ‘without penalty or loss of benefits’. 2
Trialists have raised concerns about the management of participation change for many years, often in the context of preventing missing data21–24 but also highlighting deficiencies with researcher training, 25 trial reporting 26 and information given to potential participants.27,28 We are not aware of a single, comprehensive guidance resource for managing participation changes across trial design, conduct and analysis, that addresses real-life complexity and maximally promotes participants’ interests and trial integrity.
Motivated by this lack of agreed standards and the resulting variability observed in practice, representatives from academic clinical trials units within the UK Clinical Research Collaboration (UKCRC) Registered Clinical Trials Unit (CTU) Network 29 formed a project called PeRSEVERE (PRincipleS for handling end-of-participation EVEnts in clinical trials REsearch). The project’s aims were (1) to develop principles, built on existing Good Clinical Practice (GCP) concepts, to guide how participation changes should be prepared for and handled across clinical trial design, setup, conduct, analysis and reporting and (2) to produce accompanying detailed guidance about implementing the principles. We report here the process we undertook to develop and seek feedback on the principles and guidance.
Methods
Figure 1 summarises our methods. During all project stages, we aimed for an inclusive and deliberative approach, allowing incorporation of new ideas if they were agreed to be within the scope of our objectives (with the exact scope explored and defined through testing new suggestions), and allowing time to understand objections, find compromises and get buy-in on the final outputs. All feedback was recorded and categorised systematically, and text changes were documented and shared across the collaboration for transparency.
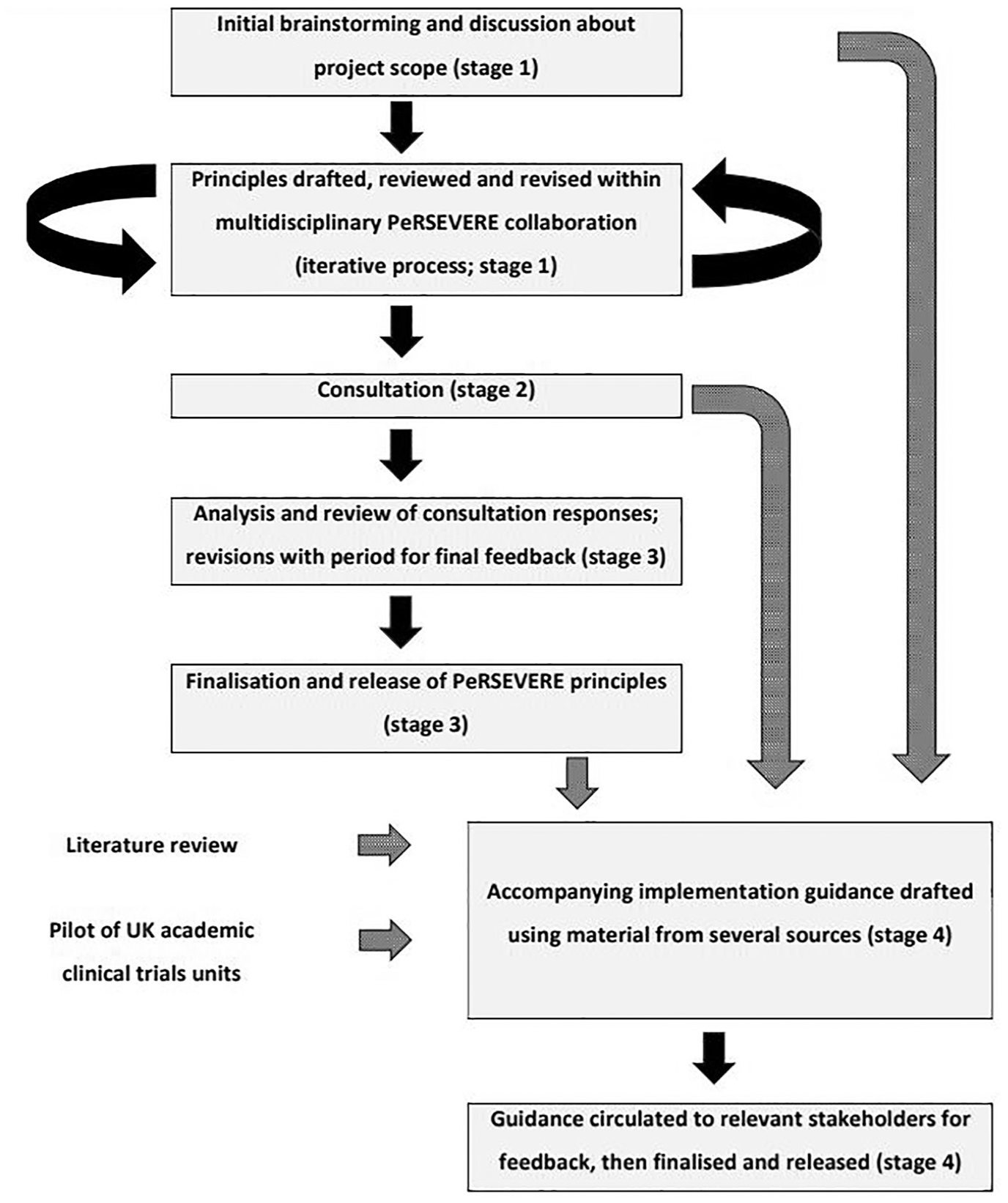
Overview of methods used to develop and consult on the PeRSEVERE principles.
Initial principle development (stage 1)
Ideas for principles were generated through a multidisciplinary meeting of representatives of 36/51 Network CTUs in October 2019, following preliminary work at the Clinical Trials Research Unit (CTRU), University of Leeds.
The principles were drafted and revised via an iterative process across seven topic-specific working groups, including statisticians, operational staff, CTU directors, research ethicists, academics and public contributors. Each working group’s leads formed a project steering group. Further detail about this initial work is available on the project website. 30
Consultation (stage 2)
We ran a cross-sectional consultation survey on our draft principles, to gather feedback from relevant interest-holders. A CROSS checklist 31 has guided our reporting of this survey (see Supplementary Materials).
Survey design and dissemination
This survey was run online via the Online Surveys platform. 32 Given the broad topic applicability, the survey was open to any interested individuals, with the only eligibility criterion excluding participation by PeRSEVERE steering group members. The survey and draft principles were provided in English.
We aimed for a balanced set of responses from a range of respondents, including people identifying as research professionals or not (e.g. public contributors), people working in industry and academia, based in different countries and with different personal characteristics. We sent the survey to research-relevant groups and individuals in the United Kingdom and internationally, with encouragement to share. Project team members sent the survey link via email, and others sharing on our behalf sent it through whichever route they considered appropriate. Given the nature of the exercise, we did not calculate a sample size but aimed for several hundred responses to give substantial feedback. Five Leeds CTRU staff unconnected to PeRSEVERE piloted the survey.
The survey protocol and question text are available in the Supplementary Materials. Respondents were asked to rate each draft principle on a 5-point Likert-type scale (‘Strongly Agree’, ‘Agree’, ‘Not sure’, ‘Disagree’, ‘Strongly Disagree’), in terms of its clarity, feasibility to implement, acceptability and novelty compared to current practice (using a conceptual framework adapted from the lead author’s prior work). 33 Respondents were given space to suggest any important considerations unaddressed by our principles and to make any other, general comments.
Respondents could choose to provide feedback on just the principles’‘key messages’ and skip groups of principles to save time. After the main survey questions, we asked respondents for information about themselves, including personal characteristics (e.g. age and ethnicity) and information on their experience with research (based on questions and categories used in steering group members’ previous projects). During stage 1, we had devised suggested, improved terminology to support clearer communication about participation changes. Our survey asked respondents for views on these suggestions.
Individuals were required to indicate their informed consent to participate before contributing. The survey requested no identifiable data.
We obtained ethical approval from the University of Leeds School of Medicine Research Ethics Committee (reference MREC 20-060) before launching the survey.
Analysing survey responses
Analysis was primarily conducted by the project lead (W.J.C.) using Microsoft Excel. During analysis, any internal inconsistencies in the data were highlighted and suitable actions agreed upon within the steering group. Missing data were included in descriptive statistics, except where a quantitative response was missing but related free text responses unambiguously indicated an answer (e.g. ‘I completely agree’).
Per-principle quantitative questions were analysed descriptively by coding the responses 1–5, with 5 being ‘strongly agree’. We summarised responses by respondents’ self-identification as primarily research professional or not. Demographic questions were summarised descriptively. We conducted exploratory analysis of different respondent groups’ opinions.
All free text responses were coded by the project lead, broadly using a framework method 34 but remaining ‘close to the raw data’, 35 that is, retaining information on respondents’ feedback, without drawing comments into too few themes. Coding of principle feedback questions was first deductive, recording whether each comment was about clarity, feasibility, acceptability, novelty or something else. All comment responses (from all open questions across the survey) were then coded inductively, starting with granular categorisation based on each comment’s content, then combining these granular categories where there was sufficient overlap. Finally, we recorded whether each comment suggested a change to the existing principles. Coding was double-checked by another steering group member (R.L. or L.C.-H.) for a randomly selected 10% of responses.
We agreed in advance how the responses should affect the draft PeRSEVERE principles. This included a rule that if the median responses to the questions about clarity and acceptability indicated at least ‘agreement’ (i.e. a median of at least 4/5) within each of the primarily professional and non-professional groups, then the current principle wording would not need substantial change. If this threshold was not met, we would use respondents’ feedback to agree on suitable updates. Lack of agreement with the feasibility and novelty questions without concerns about clarity and acceptability would not automatically imply wording changes. Instead, these lower scores would be considered in the interpretation and dissemination of our results, including in the planned implementation guidance. We agreed to consider all feedback, regardless of how many respondents made a given suggestion or the overall levels of support for the related principle. We agreed on a conservative approach to making changes to avoid overemphasising new feedback.
Other feedback during the consultation period
Feedback received via other routes (e.g. verbally after project presentations given during the consultation period) was documented, categorised and evaluated alongside survey feedback.
Finalising the principles (stage 3)
Following survey analysis, the project lead suggested responses to all survey feedback that implied we might amend a principle. Before finalisation, the suggested changes, and proposed final principles, were shared with the steering group. A summary of the significant proposed changes was sent to members of the wider PeRSEVERE collaboration and project mailing lists (which included some survey respondents).
Implementation guidance (stage 4)
The project lead drafted guidance for implementing the principles using suggestions made during stage 1, responses to the consultation, approaches suggested or implied in the PeRSEVERE principles themselves and evidence from non-systematic literature reviews. We also conducted a piloting exercise within the UKCRC Registered CTU Network. For this exercise, we asked volunteering CTUs to review the PeRSEVERE principles and comment on the extent to which they currently follow each principle (on a 5-point scale from ‘very closely’ to ‘not … at all’), how they currently follow the principles and how they might amend current practices to follow them more closely.
Before finalisation, the drafted implementation guidance was circulated for feedback to the PeRSEVERE steering group, representatives of the piloting CTUs, members of the stage 1 working groups and members of relevant Registered CTU Network operations groups. 36
Patient and public involvement
Public contributors were involved in PeRSEVERE from stage 1 onwards, to ensure a strong non-researcher presence in steering the project, given its considerable potential implications for patients and the public. At most there were seven contributors on the project steering group, who also met periodically as a standalone public contributor group. More information on the involvement activities, and a completed GRIPP2 checklist, 37 are available as Supplementary Materials.
Results
Initial principle development (stage 1)
Initial development resulted in 16 principles in four domains: ‘overarching’ principles (underpinning the overall approach; coded with an ‘O’, e.g. principle O1), ‘study development and participant information’ (‘D’), ‘data management and monitoring’ (‘M’) and ‘study reporting’ (‘R’).
Key amendments during stage 1 included principles on sharing trial results with participants who stop taking part and trial monitoring. 38 We also refined our intended level of detail (to address more about what should happen than how) and scope (e.g. to mostly exclude issues of biological sample storage).
Consultation (stage 2)
There were 280 consultation survey responses between 11 May and 27 August 2021. Table 1 summarises respondents’ characteristics. We had responses representing all 20 roles, 22 research areas and 18 research types in our category lists (see Supplementary Files), which we had intended to be as exhaustive as possible. One additional response was excluded as a likely duplication.
Consultation survey respondent characteristics.

Some of the percentages do not add up to 100% due to rounding.
In total, 269 respondents (96%) said they ‘totally’ or ‘mostly’ agreed with our key messages. Around 70% of respondents gave detailed feedback on the principles.
Table 2 provides median scores for each question about each drafted principle, overall and within the main respondent groups. More detail is available in the Supplementary Materials, including the numbers for each point on the five-point scale. Our prospective threshold about the responses on clarity and acceptability was met (median ≥4 in both categories, within both respondent groups), meaning it was not necessary to make major changes to the existing principles. The number of respondents disagreeing or strongly disagreeing for these two attributes was low (<2% for each attribute across all principles).
Median scores for each question for all draft principles, overall and within the main groups of respondents.
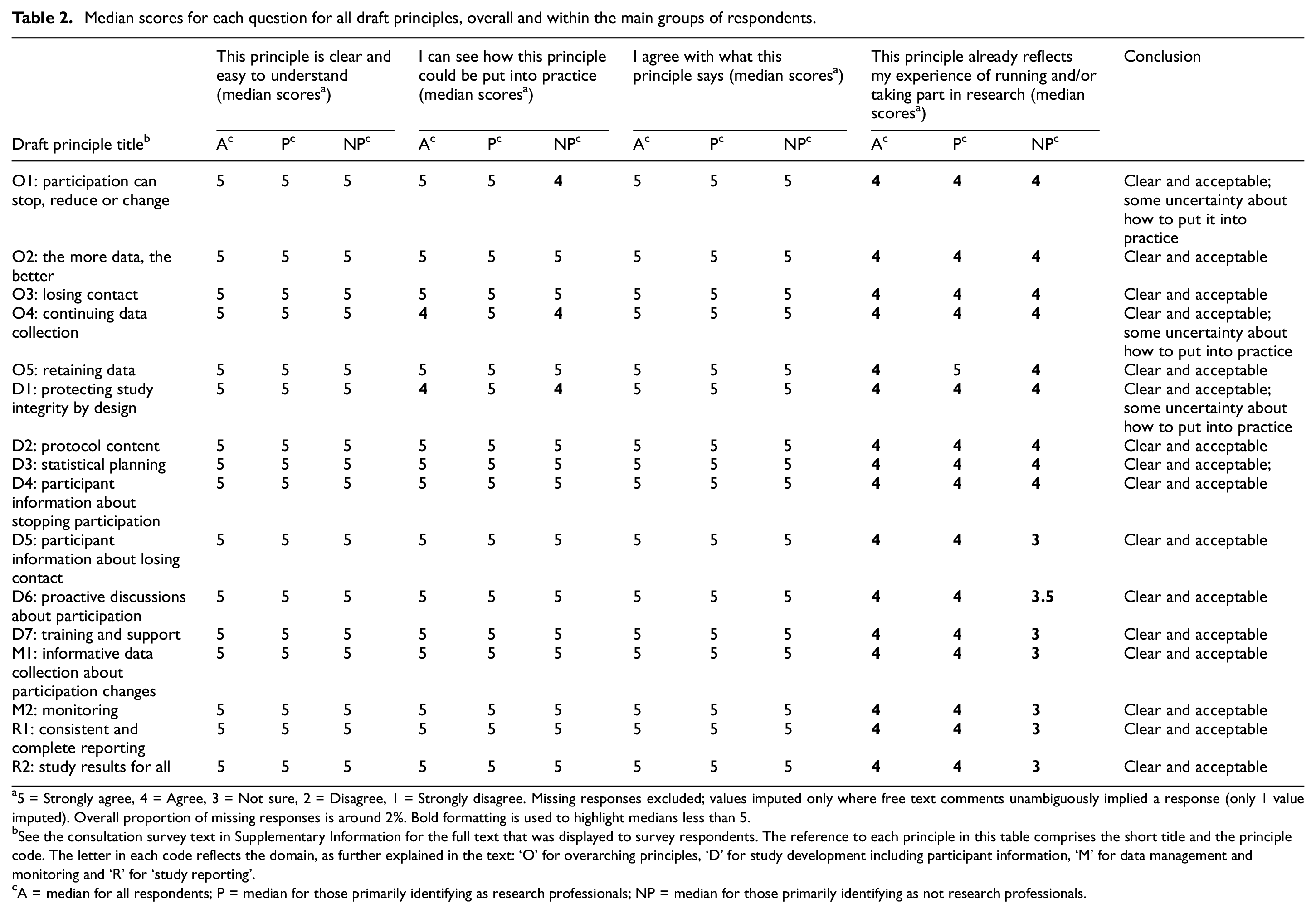
5 = Strongly agree, 4 = Agree, 3 = Not sure, 2 = Disagree, 1 = Strongly disagree. Missing responses excluded; values imputed only where free text comments unambiguously implied a response (only 1 value imputed). Overall proportion of missing responses is around 2%. Bold formatting is used to highlight medians less than 5.
See the consultation survey text in Supplementary Information for the full text that was displayed to survey respondents. The reference to each principle in this table comprises the short title and the principle code. The letter in each code reflects the domain, as further explained in the text: ‘O’ for overarching principles, ‘D’ for study development including participant information, ‘M’ for data management and monitoring and ‘R’ for ‘study reporting’.
A = median for all respondents; P = median for those primarily identifying as research professionals; NP = median for those primarily identifying as not research professionals.
Exploratory subgroup analysis suggested that groups less likely to support the principles were ethics committee members (87% saying at least ‘agree’ vs 94% for other respondents) and those with pharmaceutical industry experience (88% saying at least ‘agree’ vs 93% for other respondents). Further work would be needed to establish whether these observed differences reflect true variation between these groups’ views.
Respondents left 1071 survey comments, and there were 15 additional points of feedback outside the survey (see Supplementary Materials for full summary of free text feedback). Some recurring topics included uncertainty about how to implement the principles, worries about the risk of coercion or pressuring research participants, worries about possible negative effects on trial integrity (i.e. to do with ‘partial participation’) and difficulty applying the principles to some trial types (e.g. early phase trials).
Respondents’ views on currently used terminology (e.g. ‘withdrawn’ and ‘drop-out’) were mixed: while most respondents felt current terminology is ‘very clear’ (14%, n = 39) or ‘somewhat clear’ (39%, n = 109), a substantial minority (40%, n = 113) considered it ‘somewhat unclear’ or ‘very unclear’. Respondents felt our suggested terminology was clear and easy to understand (79% ‘strongly agree’ or ‘agree’, n = 223) and an improvement on currently used language (74% ‘strongly agree’ or ‘agree’, n = 208). Our suggestions are available in the resources section of our project website. 39
Final principles and implementation guidance (stages 3 and 4)
Major changes made to the principles after the consultation are summarised in Table 3. There are 17 finalised principles: seven in the ‘overarching’ domain, six for ‘study development and participant information’, and two each for ‘data management and monitoring’ and the renamed ‘study analysis and reporting’. The principles were made public in April 2022. The key messages from the final set of principles are shown in Table 4, with full text available in the Supplementary Materials, and on our website with explanatory guidance. 40
Summary of major changes made to draft PeRSEVERE principles following consultation survey.

The reference to each principle in this table comprises the short title and the principle code. The letter in each code reflects the domain, as further explained in the text: ‘O’ for overarching principles, ‘D’ for study development including participant information, ‘M’ for data management and monitoring and ‘R’ for ‘study reporting’.
Final PeRSEVERE principles (key messages only).
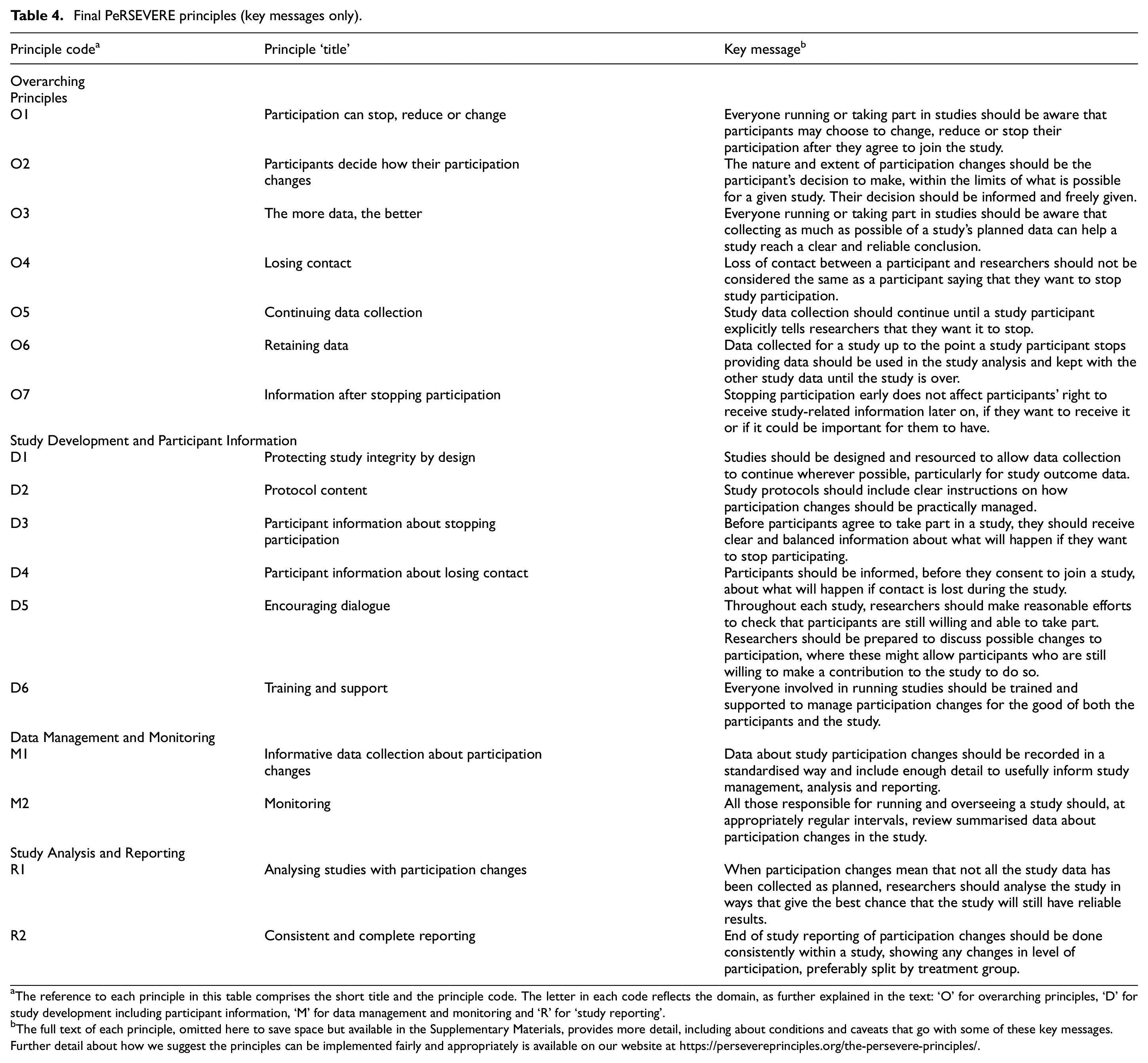
The reference to each principle in this table comprises the short title and the principle code. The letter in each code reflects the domain, as further explained in the text: ‘O’ for overarching principles, ‘D’ for study development including participant information, ‘M’ for data management and monitoring and ‘R’ for ‘study reporting’.
The full text of each principle, omitted here to save space but available in the Supplementary Materials, provides more detail, including about conditions and caveats that go with some of these key messages. Further detail about how we suggest the principles can be implemented fairly and appropriately is available on our website at https://persevereprinciples.org/the-persevere-principles/.
Eight CTUs participated in our ‘piloting’ exercise. The CTUs reported, on average, ‘somewhat closely’ following the PeRSEVERE principles. They reported ‘very closely’ following the principles about retaining data (‘O6’), analysing trials with participation changes (‘R1’) and trial reporting (‘R2’). They indicated the lowest adherence to principles about protocol content clarity (‘D2’), patient information about losing contact (‘D4’) and information after stopping participation (‘O7’).
The final implementation guidance (available on our project website) 41 contains comprehensive suggestions for applying the PeRSEVERE principles in various areas of clinical trial conduct and CTU process.
Discussion
Our final set of principles accounts for real-life complexities in how trial participation can change and provides comprehensive guidance about the management of different types of participation change. Our principles aim for maximal adherence to the GCP aims of protecting participants’ interests and trial integrity, 2 with priority given to participants’ interests where there is conflict but with detriment to trial integrity otherwise avoided. Our principles cover clinical trial design, setup, conduct, analysis and reporting and are designed to be flexible enough to apply to most or all clinical trials. These ideas may apply to other clinical research settings, although further exploration of this is needed.
Our consultation indicated good support for our draft principles from across the trials community, including research professionals and public contributors. We nonetheless used respondents’ feedback to strengthen our outputs. Many respondents felt the principles aligned somewhat with what already happens in practice, and we suggest this lends further support to our principles. This alignment could be because research professionals have tended to reach similar conclusions to ours regarding how GCP ideas apply to practical complexities of managing participation changes. However, there is still limited detailed guidance on managing participation changes in trials (although we note that content on ‘changing consent’ in the World Health Organization’s updated clinical trials guidance aligns well with our recommendations). 42 We still observe some of the same variability and uncertainty in practice that motivated the PeRSEVERE project, including on the fundamental understanding and communication of complexity in participation changes (as per our principle ‘O1’). We hope our project will replace reliance on individuals’ judgement with an established, agreed best practice.
This work has provided an opportunity to harmonise thinking across different interest-holding groups, which might otherwise have reached slightly different conclusions. Some previous publications have relied on positions defined in the PeRSEVERE principles in their methods or conclusions. For example, prior publications have mentioned that potential participants should be informed about the positive consequences of greater retention in trials.27,28 Our work strengthens the findings from previous projects by highlighting the ethical case supporting such positions and what caveats we might place around them.
The PeRSEVERE principles have potential implications for everyone associated with clinical trials, in any capacity. Table 5 highlights the specific actions that certain groups might take. Our website includes introductory briefing notes for different interest-holding groups and example scenarios illustrating the principles’ benefits (see example in the Supplementary Materials).
Suggested steps different (non-mutually exclusive) interest-holding groups might take in response to the PeRSEVERE principles.
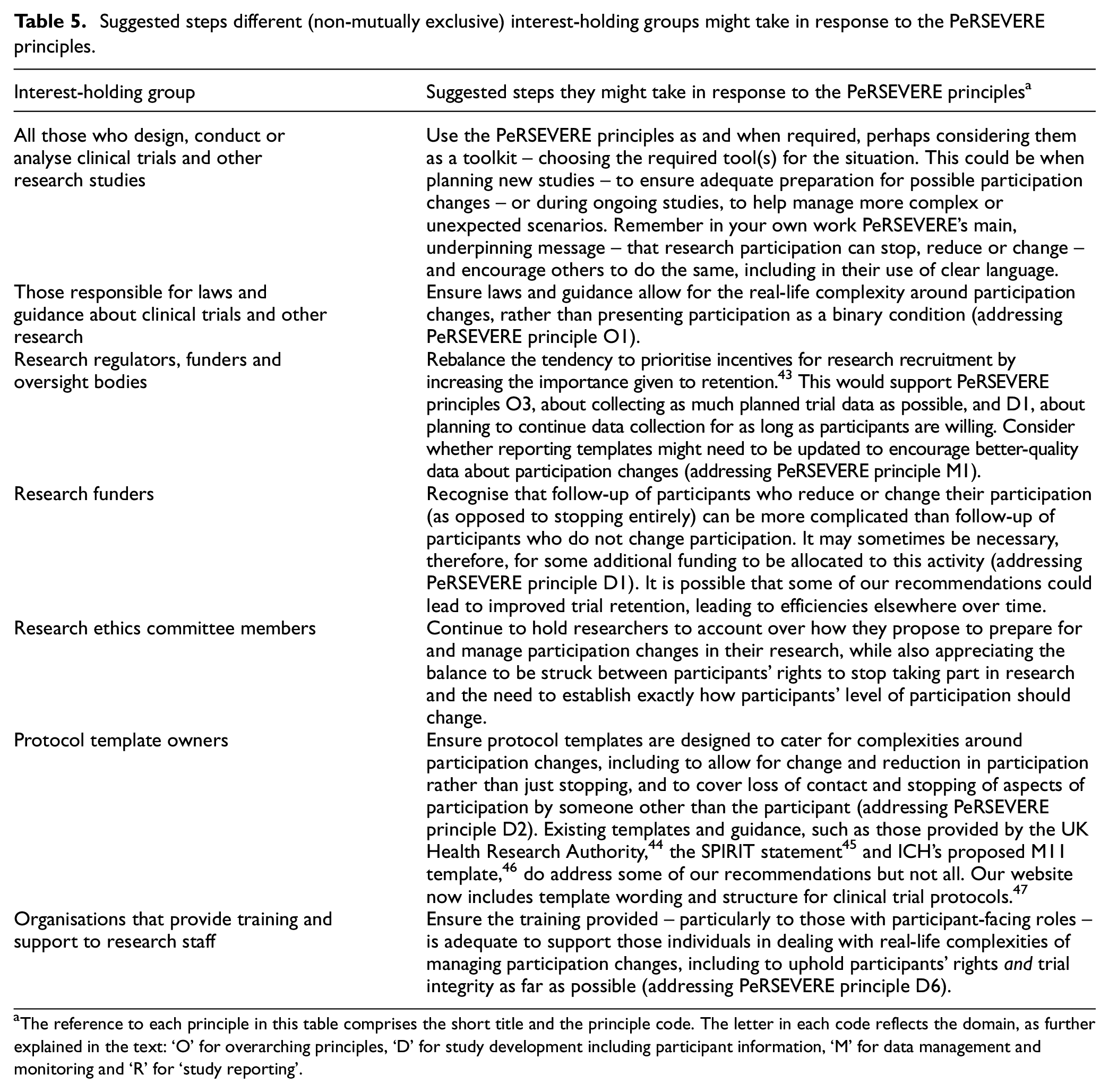
The reference to each principle in this table comprises the short title and the principle code. The letter in each code reflects the domain, as further explained in the text: ‘O’ for overarching principles, ‘D’ for study development including participant information, ‘M’ for data management and monitoring and ‘R’ for ‘study reporting’.
Recurring ideas
Some recurring ideas permeate the PeRSEVERE principles. First is the simple need to acknowledge (as per our principle ‘O1’ and as similarly noted by others, although mostly in contexts other than clinical trials)48–52 that trial participation changes can be complex, and everyone – including trial participants, if possible – should be aware of this potential complexity. Use of clear language that describes how participation has changed in any given situation should follow from this acknowledgement.
A potentially more challenging idea is that of ‘presumed ongoing consent’ in some limited cases, meaning trial activities continue until participants specifically say they want those to stop. For example, if a participant says they want to stop attending trial-specific clinic visits, then those should stop, but data collection from routine data sources can continue (if the data are relevant to the trial outcomes) unless and until the participant says they want it to stop. Some consultation respondents raised concerns about principles that rely on this presumed ongoing consent idea, including around fairness and transparency from participants’ perspectives. While we acknowledge these concerns, we suggest the approach can be a suitable way to find a balance between recognising the consent previously given, respecting the update to that consent and not harming research objectives unnecessarily. Presumed ongoing consent comes with several conditions, which somewhat resemble previous recommendations around how to justifiably treat participants’ lack of ‘dissent’ as ongoing willingness. 53 This includes being clear to potential participants about such approaches before they agree to join the trial (i.e. that they will need to specifically say they want the affected activities to stop, rather than it being implied), and expecting research staff to make reasonable efforts to establish how participants want their participation to change. In theory, it might be rare to need to rely on presumed ongoing consent, if trial processes are adequate to fully elicit participants’ wishes. However, where justified, we suggest this approach should be applied for the benefit of research integrity and therefore future patients.
A final recurring idea is about competing priorities. This is already addressed by the GCP principle that prioritises participants’ interests over those of science and society. 2 Another potential conflict can arise between participants’ freedom to stop taking part without restriction and the importance of participants’ level of participation not changing more than dictated by their wishes (and therefore, the need for research staff to know what participants’ wishes are). On one hand, we should avoid the assumption that any indication of doubt by a participant means all aspects of participation must stop. On the other hand, processes aimed at establishing how a participant’s level of participation should change cannot be a barrier to them ending their participation.52–54 For example, it is not ethical to insist that participants complete a form to indicate their wishes before they stop taking part. We recognise from our consultation that some may not agree with our recommendations and feel the existing right to withdraw consent is at risk of being diluted. This is not the intention behind the PeRSEVERE principles, and we stop short of echoing suggestions to limit participants’ rights to withdraw consent in trials or other contexts.55–58 We fully accept there will be different views on where the fulcrum should lie in striking this sort of balance, but we hope that all can at least accept that there is a balance to strike. In other words, where a participant’s level of participation will change because their wishes have changed, reducing that level more than is justified by those wishes is bad for trials and for participants.
Strengths and limitations
Our deliberative, inclusive process, incorporating contributions from several hundred individuals with different backgrounds and experiences, has helped address our aim of developing comprehensive, acceptable recommendations on this complex topic. Although we recognise that formal consensus methodology may have produced different results, we were not aiming to demonstrate consensus, only to ensure our outputs were acceptable to the clinical trials community as far as possible.
We did not conduct a systematic review for existing recommendations on this topic. However, our collective knowledge of the existing literature, and the results of a related scoping review, 59 give assurances that we have a relatively complete view of the literature. It was not feasible for us to conduct more than one consultation round, although we did share the planned major changes to our outputs with some survey respondents via the project mailing list. Although we had some non-UK responses, these were in a small minority. A large majority of our respondents identified themselves as White ethnicity. We cannot judge whether this reflects a problem in how we conducted our survey or a wider issue of a lack of diversity in research, but in any case, it means we cannot know if individuals from other ethnic groups would have different feedback. We accept that our work originates in the United Kingdom and so may not apply in the same way in other jurisdictions, cultures or healthcare systems. Further work is therefore needed to assess views on managing participation changes in a more diverse range of people, contexts, countries and cultural settings. Further work to generate evidence on how best to address our principles (e.g. standardised participant information sheet wording or data collection standards) would also be worthwhile, as would testing the effects of adherence to our principles on, for example, the availability of trial outcome data.
Conclusion
We carried out a deliberative, inclusive process to establish principles for how participation changes should be managed in clinical trials. Our final principles are more comprehensive and reflective of real-life complexity than most current ethical and regulatory guidance but still align with this guidance. Our recommendations are relevant to everyone associated with clinical trials in any capacity, and we expect their implementation will lead to improved standards in clinical trials, and a better experience for participants, in trials worldwide. We acknowledge the limitations of our project’s origins and breadth of input, and we encourage others to explore the application of these ideas in other settings and to generate empirical evidence to support best practice in this area.
Supplemental Material
sj-docx-1-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-docx-1-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-docx-2-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-docx-2-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-docx-3-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-docx-3-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-docx-4-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-docx-4-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-docx-5-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-docx-5-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-pdf-6-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-pdf-6-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-pdf-7-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-pdf-7-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Supplemental Material
sj-pdf-8-ctj-10.1177_17407745251344524 – Supplemental material for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project
Supplemental material, sj-pdf-8-ctj-10.1177_17407745251344524 for Standardising management of consent withdrawal and other clinical trial participation changes: The UKCRC Registered Clinical Trials Unit Network’s PeRSEVERE project by William J Cragg, Laura Clifton-Hadley, Jeremy Dearling, Susan J Dutton, Katie Gillies, Pollyanna Hardy, Daniel Hind, Søren Holm, Kerenza Hood, Anna Kearney, Rebecca Lewis, Sarah Markham, Lauren Moreau, Tra My Pham, Amanda Roberts, Sharon Ruddock, Mirjana Sirovica, Ratna Sohanpal, Puvan Tharmanathan and Rejina Verghis in Clinical Trials
Footnotes
Acknowledgements
We would like to thank and acknowledge the contributions of: other current and former project steering group members who are not authors on this paper, including Rachel Hobson, Joanne Milton, and Helen Moore; previous members of the patient group Sheena Hussain, Deb Morris and Kate Robinson; members of the wider PeRSEVERE collaborative group; all those who took time to provide feedback in the consultation survey and by other means; staff at the Leeds Clinical Trials Research Unit who helped to pilot the survey (Bethan Copsey, Sharon Jackson, Chris Linsley, Lynne Strutt and Jonathan Turner) and others involved initial discussions on this project; all staff at UKCRC Registered Clinical Trials Units who contributed to the piloting work. Finally, we would like to thank Gill Booth and Julia Brown for supporting the project in its initial stages, and Louise Williams, Helen Evans and Karen Griffiths of the UKCRC Registered CTU Network Secretariat for all the assistance during the project.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by the National Institute for Health and Care Research (NIHR) CTU Support Funding scheme. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. Early work on this project was supported by a Cancer Research UK Core CTU infrastructure grant (reference A24929). The funders had no role in the survey design, implementation or analysis. TMP was supported by the Medical Research Council (grant no. MC_UU_00004/07).
ORCID iDs
Data sharing
Data supporting this work are available on reasonable request. All requests will be reviewed by relevant individuals, based on the principles of a controlled access approach. Requests to access data should be made to CTRU-DataAccess@leeds.ac.uk in the first instance.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.