
Abstract
Background
Comparative effectiveness research is meant to determine which commonly employed medical interventions are most beneficial, least harmful, and/or most costly in a real-world setting. While the objectives for comparative effectiveness research are clear, the field has failed to develop either a uniform definition of comparative effectiveness research or an appropriate set of recommendations to provide standards for the design of critical care comparative effectiveness research trials, spurring controversy in recent years. The insertion of non-representative control and/or comparator arm subjects into critical care comparative effectiveness research trials can threaten trial subjects’ safety. Nonetheless, the broader scientific community does not always appreciate the importance of defining and maintaining critical care practices during a trial, especially when vulnerable, critically ill populations are studied. Consequently, critical care comparative effectiveness research trials sometimes lack properly constructed control or active comparator arms altogether and/or suffer from the inclusion of “unusual critical care” that may adversely affect groups enrolled in one or more arms. This oversight has led to critical care comparative effectiveness research trial designs that impair informed consent, confound interpretation of trial results, and increase the risk of harm for trial participants.
Methods/Examples
We propose a novel approach to performing critical care comparative effectiveness research trials that mandates the documentation of critical care practices prior to trial initiation. We also classify the most common types of critical care comparative effectiveness research trials, as well as the most frequent errors in trial design. We present examples of these design flaws drawn from past and recently published trials as well as examples of trials that avoided those errors. Finally, we summarize strategies employed successfully in well-designed trials, in hopes of suggesting a comprehensive standard for the field.
Conclusion
Flawed critical care comparative effectiveness research trial designs can lead to unsound trial conclusions, compromise informed consent, and increase risks to research subjects, undermining the major goal of comparative effectiveness research: to inform current practice. Well-constructed control and comparator arms comprise indispensable elements of critical care comparative effectiveness research trials, key to improving the trials’ safety and to generating trial results likely to improve patient outcomes in clinical practice.
Keywords
Introduction
Poorly constructed control arms or inappropriate active comparators in critical care comparative effectiveness research (CER) trials have drawn challenges and critiques for over two decades.1–7 Failure to resolve this problem stems in part from a widely held assumption that CER trials pose little or no risk to subjects because they study routinely used clinical interventions.2,8–10 This belief has led ethicists and trialists evaluating clinical trials to wrongly advise investigators, institutional review boards, safety monitoring committees, and research subjects that care as studied in some critical care CER trials will not differ substantially from care administered outside of a trial setting.2,8,9,11 Patient-Centered Outcomes Research Institute, an influential arbiter of CER policies, methodology standards states “usual care … groups should … represent legitimate and coherent clinical options.” 12 To that point, critical care CER trials commonly examine life-sustaining therapies in critically ill subjects, and deviations from “legitimate”“coherent” usual practices may yield care that is unsafe and research conclusions that are uninformative.2,7,13–19 In routine clinical practice, life-sustaining therapies are often dose-adjusted or limited to patient subsets based on clinical criteria such as history, pathophysiology, or severity of illness.2,15 Furthermore, some interventions are titrated to effect for patient safety throughout the course of critical illness. Disruption or distortion of these relationships through randomization may result in potentially hazardous “unusual care” that subjects are unlikely to receive outside of the trial.2,3,7,11,13,20
The definition of CER is intentionally broad. The Institute of Medicine defines CER as “the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition or to improve the delivery of care.” 21 The Patient Protection and Affordable Care Act describes CER as “evaluating and comparing health outcomes and the clinical effectiveness, risks, and benefits of two or more medical treatments.” 22 While these definitions emphasize the clarity CER can bring to medical practice, neither of these definitions address the hidden issues plaguing CER trial design that can confound studies and harm research subjects. Applying these non-granular CER definitions to critically ill, vulnerable subjects allows for the creation of flawed comparator groups that can undermine the safety and interpretability of these trials.2,7,11,13–19 Misconceptions about the risk profile of CER trials and the lack of preconditions guiding the selection of appropriate comparator arms have undermined a substantial number of critical care CER trials.11,13
To understand the scope of this problem, we reviewed 25 critical care CER trials published in three high-impact medical journals between 2019 and 2020. 11 Of the trials studied, eight failed to incorporate designated control and/or active comparator arms representative of contemporaneous usual critical care practices. It appears that this type of design weakness is widespread if little appreciated. This study intends to raise awareness of this issue and develop mitigating methodology that will safeguard research subjects while preventing the adoption of improperly or inadequately tested alterations to clinical practice.
Methods/examples
Definition of usual critical care
For this analysis, we define “usual critical care,” as it applies to CER, as “management consistent with contemporary practices and interventions that would have been received routinely outside of the trial.” Deviation from common practices in control or active comparator arms after randomization in a trial thus constitutes “unusual critical care.” To determine what constitutes “usual critical care,” we consider not only the therapy or intervention itself but also the specific way it would be administered to each patient. While clinical medicine varies considerably in clinical practice, not all the variation is random. Non-random variability in care is typically driven by patient characteristics, clinical factors, and disease severity, and it is vulnerable to disruption by randomization. By failing to offer differential care to patients with well-recognized differences, trial authors create practice misalignments that are at once unconventional (considered “unusual care” or not “usual critical care”) and potentially unsafe. To avoid this error, trial authors must study objective contemporary data of usual critical care across participating institutions prior to trial design and enrollment. The strongest sources of such data include investigator-initiated surveys and observational studies. Prevailing local or national guidelines should also be examined to characterize current critical care practices; however, guidelines alone cannot serve as a substitute for actual data if available or could be obtained. Moreover, some guidelines are built around general statements that overlook common individualization patterns at the national, regional, local, or institutional levels.2,16 Most importantly, at least one trial arm must provide usual care after randomization. If two active comparators arms are contrasted in CER, then both arms need to provide usual care after randomization. Otherwise, the results of the trial can have no direct applicability to current clinical practice. Some published studies fail to recognize major deviations from usual care and/or how care is commonly provided outside of the trial; this can also lead to a misinterpretation or application of study results. Experimental approaches that compare two new treatments but lack a contemporaneous usual care arm severely compromise any ability to determine the equivalence or superiority of either novel approach to current practice.
We found that many recent critical care CER trials clearly define and incorporate usual care into their trial designs. To examine this as stated above, we performed a systematic review of high-impact clinical trial journals and determined that 12 of 146 randomized clinical trials published in The New England Journal of Medicine from April 2019 to March 2020 met the Institute of Medicine criteria for critical care CER. 11 Six of these relied on contemporaneous data to define and incorporate current practices into trial design.23–28 Across these six trials, investigators were exacting in defining usual care. The investigators responsible for these six trials performed 19 separate studies to understand and characterize usual care. The investigators additionally cited more than 60 articles to define contemporaneous practices. 11 We further found that, in 11 out of 13 critical care CER studies published in The Lancet and Journal of the American Medical Association during the same time frame, trial authors also meticulously characterized contemporary practices a priori and rigorously replicated these practices in the design and conduct of the trial.29–39 Based on these findings, we would dispute any suggestion that it is not possible to characterize and/or implement usual care in critical care CER trials. 11
Risks of not studying usual care in CER
Critical care CER studies that lack a control arm—or are comprised of two or more active comparator arms which fail to reliably incorporate contemporaneous usual care—create the following risks: (1) Monitoring boards cannot easily determine whether an intervention is harmful or beneficial compared to usual critical care. Thus, the boards cannot reliably detect a signal for benefit or harm. For an illustrative case, see Example 10 in section “Type 2B trials.” (2) If usual critical care is not studied, it becomes nearly impossible to know if conclusions made from any comparisons made will, with assurance, improve future patient care. For an illustrative case, see Example 8 in section “Type 2B trials.” (3). Informed consent—which should clearly explain usual critical care practices and how care will differ in context of the trial—is compromised. For an illustrative case, see Example 5 in section “Type 2A trials.”
Types of CER trials and common design errors
We propose that there are at least three common types of Critical Care CER trials—Types 1, Type 2A, and Type 2B (see Table 1). Each of these types is associated with persistent, specific, and unique design errors.
Clinical trial types and the most frequent design errors and potential results.
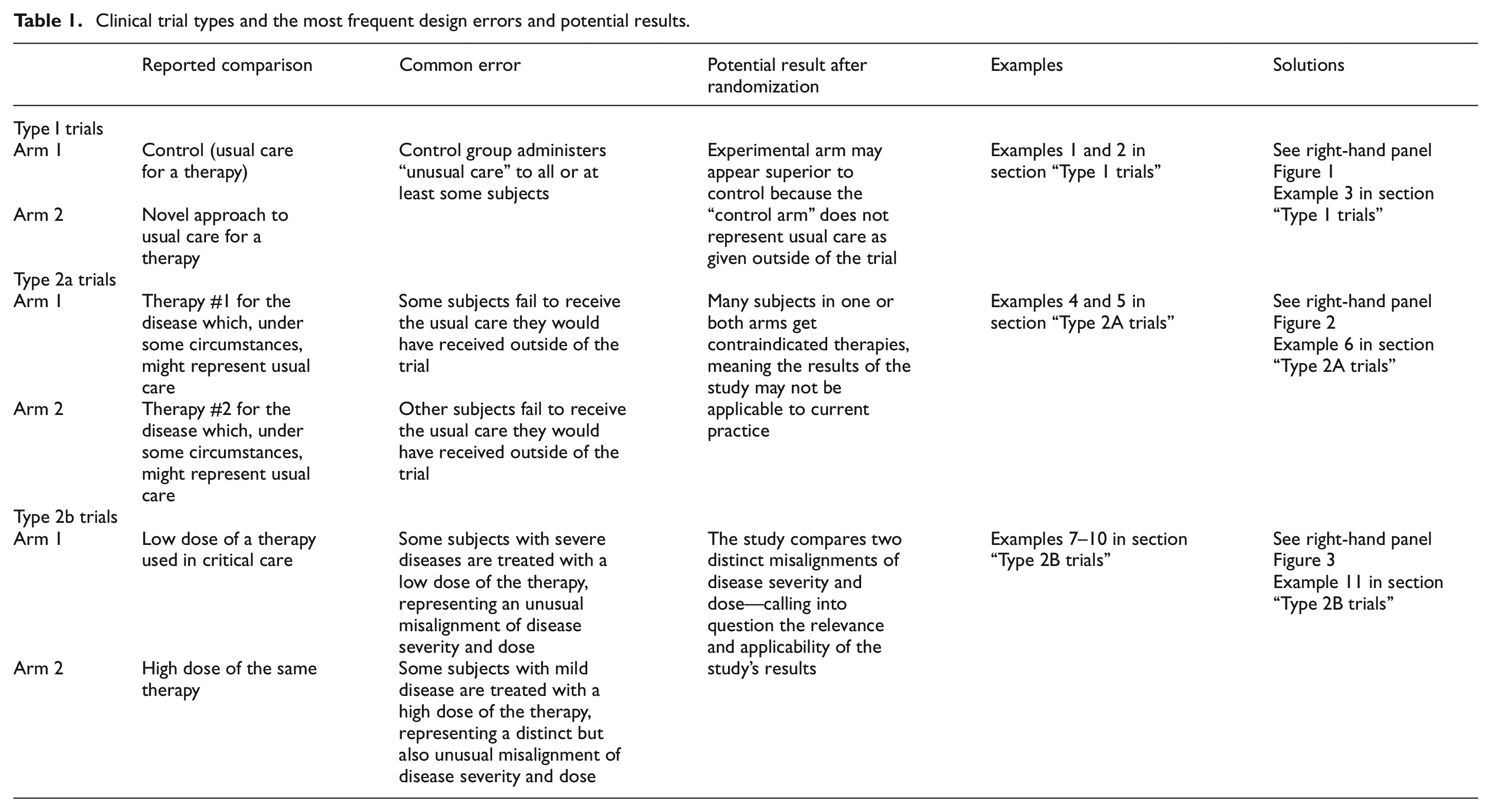
Type 1 trials
These compare a designated usual critical care control intervention to an unusual but potentially beneficial and acceptable modification of that intervention (Figure 1 and Table 1). A common error is that the trial design fails to establish usual critical care as it is practiced outside of the trial and instead provides novel, potentially unusual, critical care across both arms.
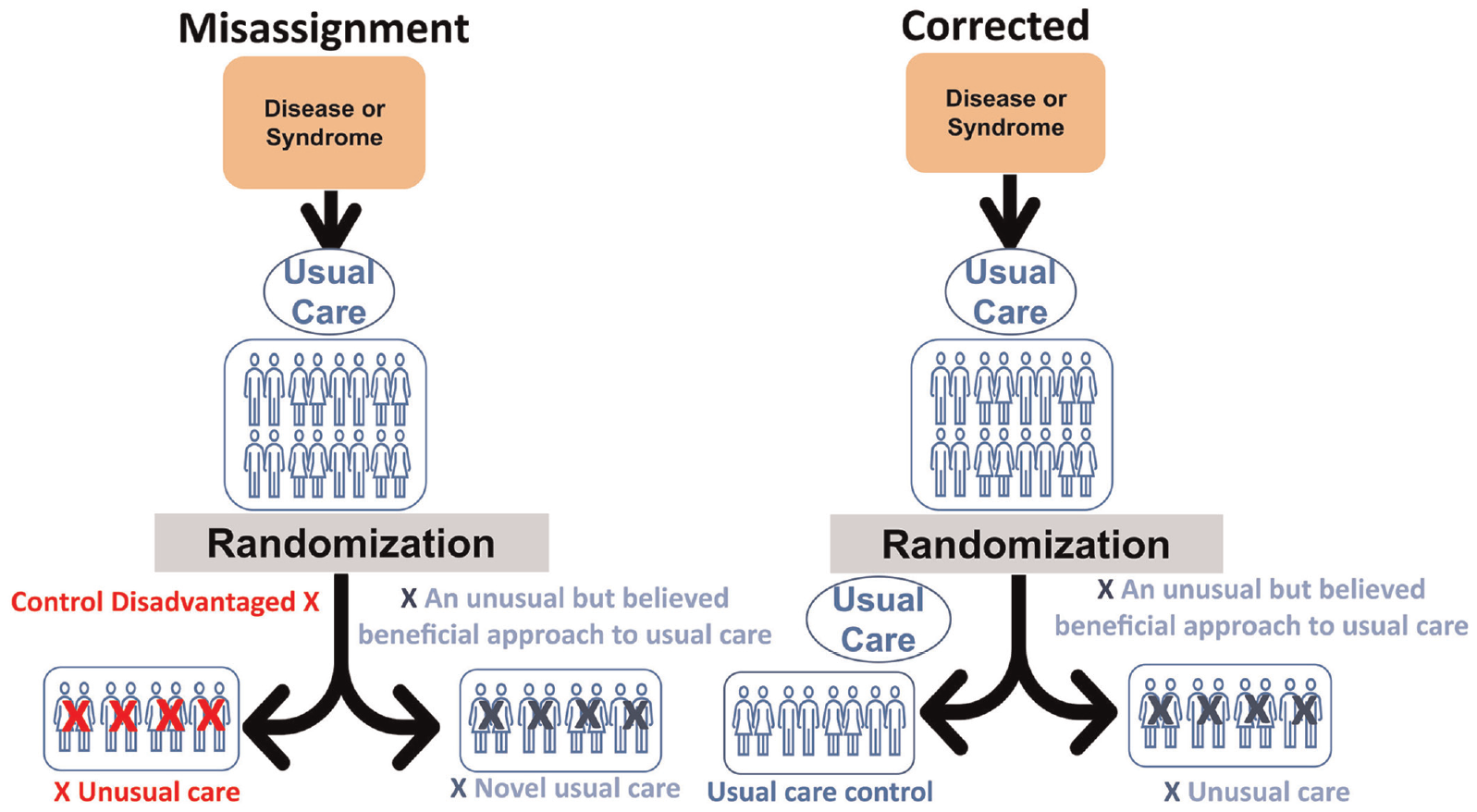
Type 1 CER—a usual critical care control compared to a novel or unusual approach to usual care believed potentially beneficial. For a Type 1 CER comparison (“usual care vs novel usual care”), a common error is the therapeutic misalignment. In this case, the error consists of disadvantaging the control arm. The trialists are comparing a novel approach toward usual care to a disadvantaged control, meaning the trialists cannot determine whether the novel therapy is beneficial or whether the disadvantaged control is harmful. Furthermore, the study’s results cannot easily advise current practice, as the trial design ensures it was never actually studied. The right-hand panel shows how trialists, by studying usual care pre-randomization and verifying it again post-randomization, can ensure the control arm still comprises usual care, eliminating the possibility of a therapeutic misalignment and making the findings readily applicable to current practice.
Example 1, Type 1 error: a recent trial compared cardiac arrest subjects treated with hypothermia to a designated “normothermia control.” 40 After randomization, subjects in the arm designated “normothermia control” with low temperatures at enrollment were warmed until their core temperature reached an assigned normothermia range. Contemporary critical care practice and guidelines did not support actively warming such patients.11,41–45 In this design, the “control” represented unusual critical care, potentially disadvantaging this arm compared to the experimental treatment and making it difficult to determine whether the hypothermia arm was beneficial or the “normothermia” arm was harmful.
Example 2, Type 1 error: a recent trial investigated whether early neuromuscular blockade in acute respiratory distress syndrome improves outcomes compared to usual care. 46 After surveying only the primary site investigators, trial authors restricted use of neuromuscular blockade in the “usual care control arm” to refractory hypoxia and/or to subjects with plateau pressures above 32 cm H2O that persisted for at least 10 min despite increasing sedation and decreasing positive-end expiratory pressure and tidal volume. However, this did not constitute current practice: data from a large survey, as well as most published observational data available at the time, conclusively showed that clinicians did not commonly restrict neuromuscular blockade in this manner. Instead, clinicians administered neuromuscular blockade most commonly for other clinical indications, such as ventilator asynchrony.11,47–50
Example 3, Type 1 error avoided: in a trial comparing sedation practices of dexmedetomidine versus usual care in intubated mechanically ventilated subjects, investigators diligently determined usual care a priori to avoid this Type 1 error.11,26 Three observational studies published by the investigators, and another referenced in the trial and protocol, helped define and support their design of the usual care arm that was employed.51–54 A prospective observational study found that dexmedetomidine was used in only 7.6% of patients in the first 48 h following intubation. 51 Therefore, the control arm was designed to replicate usual care by only discouraging dexmedetomidine use in the control arm but fully permitting dexmedetomidine if additional sedation was deemed necessary by treating clinicians at the bedside.
Type 2A trials
These compare two or more different active comparator treatments, all of which are defined as usual critical care (Figure 2 and Table 1). The common error is the therapeutic misalignment occurs when randomization leads to subjects within a trial being administered therapeutics and treatments which clinical characteristics would usually preclude them from receiving during routine care outside of the trial. 15 In the case of critical care Type 2A error, the two or more treatments being compared are different from each other but treat the same disease. However, each treatment during contemporaneous common practice is typically administered to patients based on their distinct clinical characteristics. This is because each approach may be associated with perceived risks and benefits that differ across different patient subgroups. Therapeutic misalignments result when investigators fail to consider these practice/patient relationships and decouple them through randomization—thus causing patient subgroups across one or more arms to receive therapies or interventions that would be considered inappropriate outside of the trial setting, meaning investigators have administered unusual care to trial subjects. Any trial that only compares two arms—both constituting unusual care—cannot yield results that reliably inform current critical care practice.2,15 Current practice might be superior to the two unusual care arms but remain unknown due to its omission from the trial design.
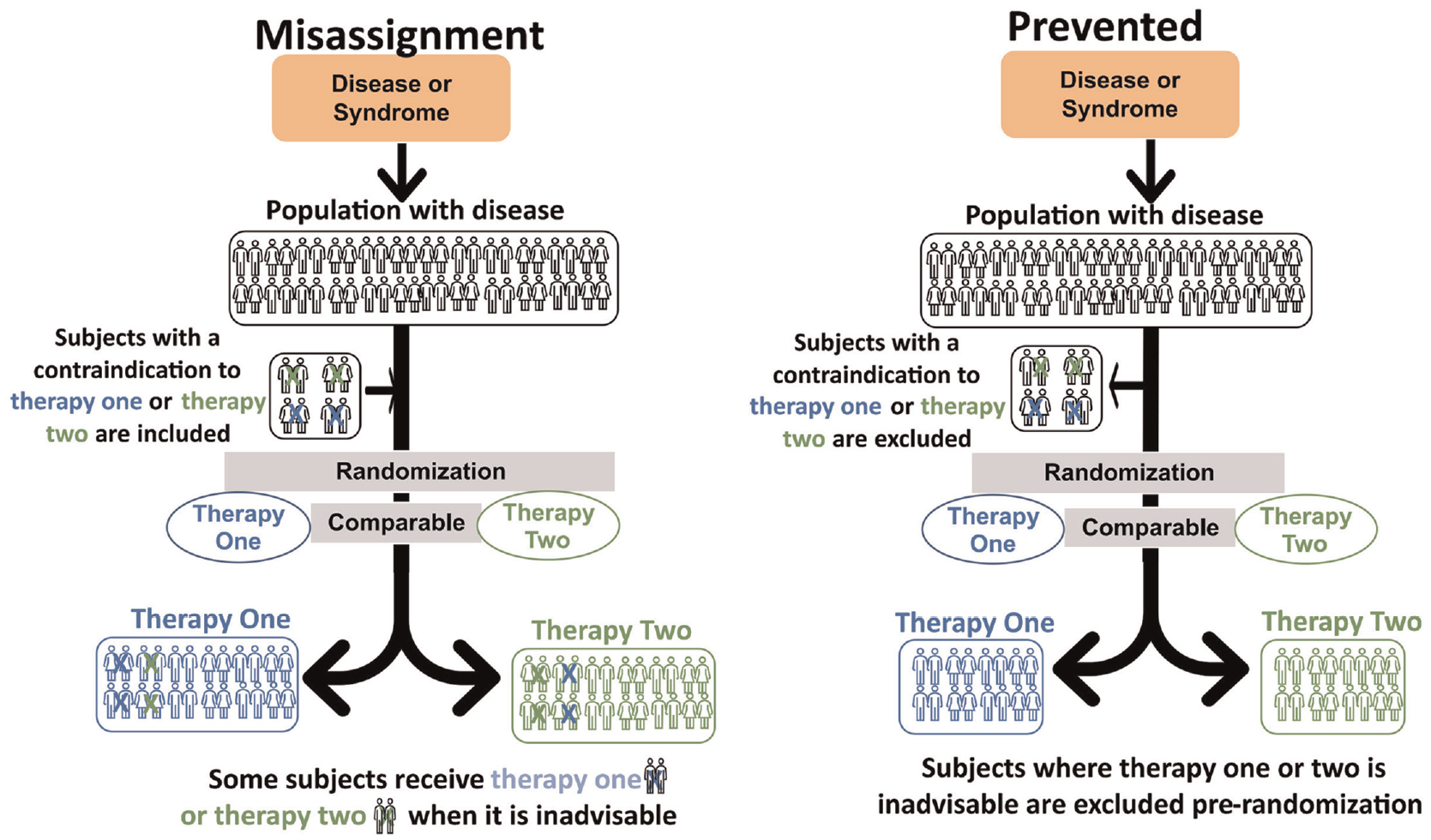
Type 2A CER—two active comparator arms which vary categorically from each other compared. For Type 2A CER, the panel on the left shows a therapeutic misalignment. In both arms, post-randomization, some subjects received an inadvisable treatment. This comparison has limited clinical meaning. Furthermore, trialists have limited ability to advise current practice as it was never studied. The figure on the right shows how this error can be avoided if trialists, prior to randomization, exclude subjects for whom either therapy is inadvisable. The correction in Type 1 can also be used to help prevent this error.
Example 4, Type 2A error: in a recent trial studying the acute management of patients with status epilepticus, trial authors randomly assigned a broad, heterogeneous group of subjects to receive one of the three antiepileptic drugs. 55 In routine practice, physicians select such treatment after considering which of the three drugs patients are already receiving, as well as their compliance history, age, comorbidities, and underlying conditions.13,56–58 But in this trial, the authors failed to account for any of these factors for the majority of trial subjects and instead randomized heterogeneous subjects to receive any one of the three trial drugs regardless of their histories or of the dictates of usual practice. In approximately 10%–15% of subjects, the etiology of the episode of status was withdrawal of or non-compliance with taking their home antiepileptic drugs. 55 If poor compliance with taking home antiepileptic drugs is suspected, the preferred treatment after benzodiazepine administration would be to administer additional doses of their maintenance antiepileptic drugs. 57 Arbitrarily selecting an antiepileptic drug in an individual who was in status because of missed doses instead of giving additional doses of their maintenance drug known to have previously provided seizure control would constitute unusual care and potentially delayed effective treatment. Despite this deviation from usual care, the Food and Drug Administration allowed a waiver of informed consent. 55 Ultimately, the authors compared three Food and Drug Administration approved drugs that were randomly assigned in contradistinction to usual practices. As such, the ability to inform current practice was compromised since it was never studied.
Example 5, Type 2A error: in a trial investigating target oxygen saturation range, neonates born at less than 28 weeks’ gestation were randomly assigned to the lower or upper half of the American Academy of Pediatrics’ recommended range of oxygenation. 59 Relying solely on this range, the trial authors failed to consider or incorporate previously published available data outlining “current” practices.16,60 The study as performed ignored the common practice of neonatologists to almost always set the upper limit of the oxygen saturation range at or above 92%, thus allowing bedside caregivers to err on the side of adequately oxygenating all neonates. While the lower limit of the range was highly variable,2,13,16,60 nurses were found to routinely skew oxygen delivery toward the upper end of target ranges to avoid hypoxia.16,60 The upper limit for the lower oxygen saturation range studied (89%) in the trial was rarely, if ever, administered in clinical practice. 16 This limited the maximum level of oxygen that can be delivered to only low dosages by restricting the upper limit of the overall range. Delivering low levels of oxygen saturation can be extremely harmful to neonatal subjects, increasing the risk of necrotizing enterocolitis and death.16,61 The consent documents for this trial did not include this risk. The documents misled parents by stating that there was no increased risk associated with taking part in the study and that, in the two arms studied, neonates would receive “routine” or “standard” care.16,20,62 Furthermore, within a year after publication in The New England Journal of Medicine, this trial resulted in a 3-year long controversy in the lay and scientific press over the adequacy of the consent documents explanation of risks in consent documents provided to the parents.2,9,16,59,63,64 Defenders of the trial argued that since both arms are usual care, informed consent wasn’t even necessary.9,10 This controversy abruptly ended once it was shown that one of the two oxygen ranges studied was lower than usual care, had increased risks and was rarely if ever used in neonatal intensive care units. 16 This trial illustrates how important it is to understand contemporary practices and not just guidelines in developing well-designed critical care CER trials.
Example 6, Type 2A error avoided: usual care was effectively incorporated in a trial comparing whether high-flow nasal cannula therapy in premature infants with respiratory distress was non-inferior to nasal continuous positive airway pressure.11,25 Prior to commencement, the investigators conducted a survey that indicated most healthcare providers used nasal continuous positive airway pressure and only a few centers additionally used high-flow nasal cannula therapy in infants. 65 Therefore, to maintain usual care, enrollment was restricted to centers using nasal continuous positive airway pressure alone. Given this restriction, investigators did not have to determine whether other factors influenced the choice of treatment and did not need to incorporate such factors into the trial design. Focusing enrollment on the chosen centers made it possible to draw firm conclusions for institutions that primarily used nasal continuous positive airway pressure and could therefore easily apply to common practice.
Type 2B trials
Compares different doses or levels of the same treatment that is commonly titrated over a range based on patient characteristics during routine care (Figure 3 and Table 1). The common error is the therapeutic misalignment between dose or level and disease severity. Many critical care interventions are adjusted to individual patient needs, reflecting differences in patient-level characteristics that can evolve over time during critical illness. Errors result when trial authors conduct randomization that misaligns treatment dose or intensity with the needs of individual patients, subjecting them to care different from what they would receive outside of the trial. Instead of preserving routine titration practices, trial authors instead randomize subjects to fixed levels of the same treatment. 15 In general, there is a known significant relationship between a treatment and clinical characteristics which are based on physiologic outcomes that must be preserved or the subjects will be harmed. Therefore, randomizing subjects to two fixed and widely separated treatment doses of a routinely titrated therapy—irrespective of the severity of subjects’ illness or need—means that subgroups in both arms will receive different, non-comparable unusual care. In one arm, a subgroup of subjects with the least severe disease will be randomized to receive maximal therapy. In the other arm, a subgroup with the most severe disease will be randomized to receive minimal therapy. Comparing these two arms has limited clinical applicability.
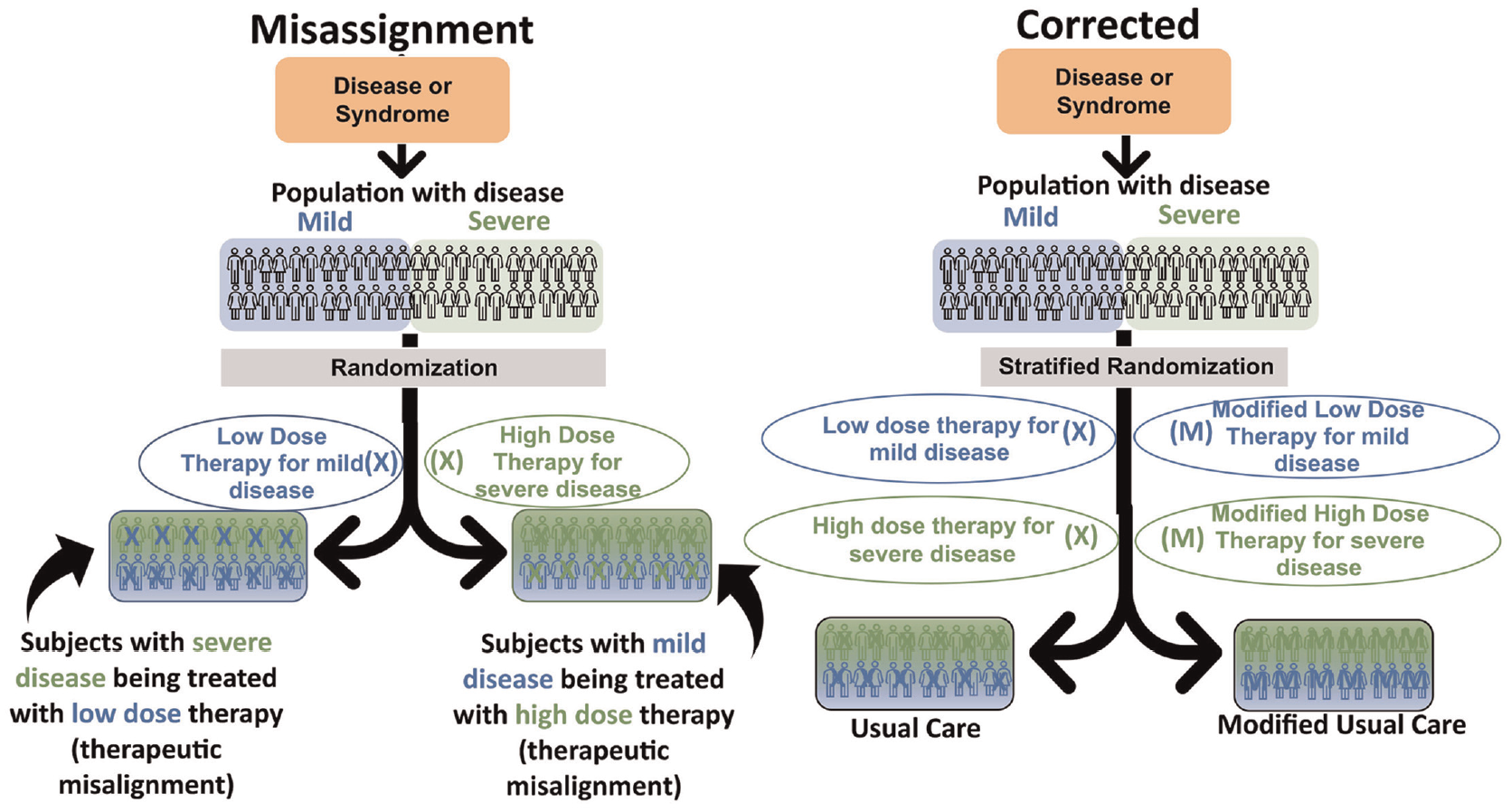
Type 2B CER—two active comparator arms which vary orthogonally (by dose) from each other compared. For Type 2B CER, a therapeutic misalignment on the left panel is shown. Therapy variation is not random in this case but is based on a clinical characteristic that is likely to alter patient outcome. Although the therapy dose covaries with severity of disease, trialists disregard this fact. This results after randomization comparing some subjects with severe disease receiving low-dose therapy to subjects with mild disease receiving high-dose therapy. This comparison has limited clinical meaning to current practice. Thus, the trialists’ ability to correctly advise practice is diminished, as it was never studied. The panel on the right shows how these misalignments can be avoided if trialists stratify randomization to maintain the current practice of adjusting dose by disease severity in one arm. In the other arm, one can modify the low-dose and high-dose therapy in a manner believed beneficial for each and stratify the randomization, so the appropriate severity disease is studied with a suitable therapy dose. It is also possible to circumvent the need for stratification and avoid misalignment by studying the experimental therapy in comparison with either only the low-dose therapy as given in current practice or the high-dose therapy as given in current practice.
Example 7, Type 2B error: this problem can be easily seen in a hypothetical trial of vasopressor therapy. In routine care, vasopressors are carefully titrated to maintain a target blood pressure in septic shock. Patients with severe septic shock will require higher doses of vasopressors compared to milder cases who may require little or no vasopressor administration. A hypothetical study can be designed where subjects are randomized to receive either high- or low-dose vasopressors regardless of the severity of shock. This produces a predictable misalignment: some subjects with severe septic shock randomized into the low-dose arm will receive insufficient therapy and have persistent hypotension. Conversely, in the high-dose vasopressor arm, there will be a subgroup of subjects with mild shock who, despite clinically requiring minimal vasopressors, receive excessive doses and become hypertensive. Therefore, the comparison between these two fixed dosing arms of this hypothetical study is a meaningless one and reveals only which practice misalignment is more harmful. 15 If prescribed dosage in clinical practice significantly covaries with severity of illness or other patient determined factors, then randomly assigning patients to two widely separated, fixed treatment regimens will likely produce qualitative interactions or different harmful effects in each arm. If this is not recognized, then whichever arm is more harmful will determine the overall outcome of the study. If presence of a qualitative interaction is not recognized, the less harmful of the two clinical scenarios could be enshrined in perpetuity in future clinical practice.
Example 8, Type 2B error: a trial of critically ill subjects studied how restrictive and liberal approaches to red blood cell transfusion affect mortality. 66 A prior survey by the investigators showed that physicians typically employ a range of hemoglobin levels to trigger transfusions and prescribed more red blood cell units as patients’ age, cardiovascular comorbidities, and severity of illness increase, consistent with contemporaneous consensus practices. 67 Despite the survey findings supported by consensus conferences at the time, 68 the trial authors did not include a usual critical care arm in which they adjusted transfusions based on individual patient characteristics. In one arm, younger, stable subjects received transfusions that were not clinically indicated and potentially harmful—while, in the other arm, older subjects at risk for cardiovascular disease did not receive transfusions that were clinically indicated.2,13–15,17 Comparison of these two arms was uninformative as it was a comparison of two different types of unusual critical care. This study could not fully inform contemporary practices as current usual care was not incorporated into the trial design. Whether either of the fixed-dose arms (both experimental) might be better than usual care, which is routinely titrated, is not known.14,15,17,68
Example 9, Type 2B error: in another trial where routinely titrated therapies were instead studied at fixed levels, the authors compared a set high versus low range of arterial blood oxygen during the treatment of acute respiratory distress syndrome. 69 In no trial arm did clinicians practice usual critical care, which involves titration of supplemental oxygen to avoid hypoxia while minimizing patients’ exposure to toxic oxygen levels.11,47,70–74 Consequently, some patients with severe disease randomized to the low fraction of inspired oxygen arm were unnecessarily kept in a state of relative hypoxia. 11 Other subjects with minimal disease randomized to the high arm of fraction of inspired oxygen were unnecessarily exposed to potentially toxic O2 levels despite their high blood oxygen levels. 11 Ultimately, this trial compared two arms in which different subgroups received unusual critical care, increasing trial subjects’ risk and significantly diminishing the usefulness of results for informing current practice. 11
Example 10, Type 2B error: in another severe respiratory failure acute respiratory distress syndrome trial, subjects were randomized to two fixed treatment strategies one with mechanical ventilation with a large breath versus the other arm which had a small volume breath (high vs low tidal volume ventilation). 75 The baseline data from this trial showed that healthcare providers typically used a range of tidal volumes pre-randomization.2,13,15,18,76 On average, the pre-randomization tidal volumes decrease as lungs became more rigid (non-compliant) or diseased.76,77 But subjects in this trial’s high tidal volume arm, designated “traditional volume,” did not receive care consistent with contemporaneous practice as defined by current mechanical ventilation data. Approximately 80% of subjects enrolled in the high tidal volume control arm saw their tidal volumes increased from pre-study baseline values as prescribed by their personal physician. 15 The death rate was much higher in the high tidal volume arm than it was for patients who met enrollment criteria but were not randomized. 76 In this design, the “traditional volume” represented unusual critical care, likely disadvantaging subjects in this arm. Moreover, the trial’s design rendered it difficult to determine whether the intervention in the low tidal volume arm was beneficial or the high “traditional volume” arm harmful.2,13,15,76,78
Example 11, Type 2B error avoided: an example of a trial maintaining usual care titration on all arms investigated if a more tightly managed routine oxygen therapy in acute respiratory failure would prevent lung injury.11,23 One arm of this trial was unrestricted usual titrated care which was determined by eight previous studies conducted by the investigators as well as four observation studies published by others.70–74,79–85 The other arm was a more conservative arm where healthcare providers more fastidiously lowered blood oxygen levels if the oxygen saturation was at acceptable levels of >91% which was consistent with current practice. The trial maintained usual titration practices in both arms; using the lowest inspired oxygen therapy that would result in a safe level of arterial oxygenation. They tested whether a more conservative strategy characterized by more aggressively lowering oxygen levels whenever possible could further limit unnecessary hyperoxia and injury.
Considerations for CER trial design
We have proposed a schema to organize critical care CER trials. Across the three types of error, the foundational flaw in trial design leading to the common errors we describe is the lack of a control or active comparator arms representing contemporaneous usual care. To safeguard against this widespread error, we suggest the following “best practices.” Step (1) Investigators should perform an in-depth determination of contemporaneous usual care practices at enrolling institutions before designing critical care CER trials. This process can include—but is not limited to—literature review, surveys, and retrospective or prospective observational studies. The investigators should define “usual care” by clearly documenting how therapies and interventions are administered, dosed, and adjusted based on disease dynamics and individual characteristics of all major subtypes of subjects. Step (2) Critical care CER trials should be designed to incorporate usual care by one or more of the following methods: for a Type 1 study, at least one arm should reasonably represent usual care as administered outside of the trial. For a Type 2 study, multiple arms of the trial must constitute usual care. Trial authors must consider and apply appropriate exclusion or inclusion criteria when designing their trials so that, after randomization, subjects enrolled in one or more arms of a trial still receive usual care. Step (3) After designing the CER trial, but prior to enrollment of subjects, trial authors should conduct a thought experiment.
In a Type 1 trial, the authors should ask whether, after randomization, the different major subtypes of subjects enrolled in the “control” arm will still receive usual care. If necessary, the authors should then modify their originally planned exclusion criteria to exclude those subjects deemed unlikely to receive usual care post-randomization. In a Type 2 trial, the authors should examine all active comparator arms to ensure that enrolled subjects will still receive usual critical care post-randomization as it is practiced for that specific population of patients. Once again, the authors must modify planned exclusion criteria—or develop additional ones—to account for those subjects deemed unlikely to receive usual care post-randomization.
Limitations
The limitations of applying our recommendations to critical care CER require mention. In this article, our focus is not on whether critical care CER trials ask clinically meaningful questions or whether the informed consent documents were adequate. Rather, we focus on whether trial designs optimized their ability to inform current practice, facilitated (or allowed) the writing of informed consent documents, and minimized risks to subjects through an understanding of usual care and capacity to adequately monitor safety. We acknowledge that there may be additional errors of trial design involving lack of or inappropriate use of usual care comparators in CER.
Conclusion
It is essential to both define and incorporate usual care when designing and conducting critical care CER trials. In some past studies, insufficient attention to these matters compromised study conclusions, patient safety, and the process of informed consent.2,11,13–19 Our proposed schema may not capture every potential type of critical care CER comparison, and we acknowledge that some of the classification types overlap. Furthermore, we understand that our recommendations could make it more difficult to recruit and enroll patients. However, we believe that the approach we outline is not only feasible but will decrease risks and generate valid study results that have real-world implications. We are confident that our proposed definitions and categorizations, as well as our description of common errors, will constitute a meaningful step toward the improvement of critical care CER trial design.
Footnotes
Acknowledgements
The authors thank Ruth Macklin and Michael Carome for their invaluable help in ensuring the accuracy and readability of the manuscript. The authors dedicate this manuscript to the memory of Jordi Mancebo, who died on 6 August 2022, at 64 years. Jordi reviewed an earlier version of the manuscript, and we can only hope it captures his passion for truth and dedication to patients.
Author contributions
C.N. and H.K. contributed to conceptualization. C.N., H.G.K., and V.J.F. contributed to data curation. V.J.F. contributed to visualization. C.N., H.G.K., P.Q.E., V.J.F., I.C.-P., W.N.A., and J.W. contributed to writing. H.G.K., C.N., and V.J.F. contributed to editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH) intramural funding from the NIH Clinical Center. The work by the authors was done as part of US government–funded research; however, the opinions expressed are not necessarily those of the NIH.