
Abstract
Background
Informed consent is the cornerstone for protection of human subjects in clinical trials. However, a growing body of evidence suggests that reform of the informed consent process in the United States is needed.
Methods
The Clinical Trials Transformation Initiative conducted interviews with 25 experienced observers of the informed consent process to identify limitations and actionable recommendations for change.
Results
There was broad consensus that current practices often fail to meet the ethical obligation to inform potential research participants during the informed consent process. The most frequent single recommendation, which would affect all participants in federally regulated clinical research, was reform of the informed consent document. The interviews also identified the need for reform of clinical research review by institutional review boards, including transitioning to a single institutional review board for multi-site trials.
Conclusion
The consensus recommendations from the interviewees provide a framework for meaningful change in the informed consent process. Although some proposed changes are feasible for rapid implementation, others such as substantive reform of the informed consent document may require change in federal regulations.
Keywords
Introduction
Improving the effectiveness and efficiency of the informed consent process is a critical health policy issue. Informed consent is an essential element of ethical biomedical research, with federal regulatory requirements that are intended to ensure autonomy of the research participant’s decision to engage in research after a balanced discussion. 1 However, a growing body of evidence suggests that the informed consent process does not fully satisfy the needs of clinical research participants,2–4 with a recent survey finding that nearly 19% of prospective participants considered the informed consent document difficult to understand and that 15% were not satisfied that their questions had been answered during the consent process. 5
It has been more than 20 years since the last substantive revision of federal regulations for human subjects protection, including informed consent. In 2011, the US Department of Health and Human Services issued an Advance Notice of Proposed Rulemaking with proposed reforms. 6 Although this led some to expect that revision of human subjects protection regulations was imminent, major reform has yet to occur.
Recognizing deficiencies in the current informed consent process, the Clinical Trials Transformation Initiative (CTTI) 7 launched the Informed Consent Project. 8 Initial project activities revealed that although an extensive body of literature on informed consent exists, there is little published information examining the observations and recommendations of experts with long-standing experience with the informed consent process. Accordingly, a CTTI work group (the Expert Interviews Team) conducted interviews and analyzed information from 25 highly experienced observers of the informed consent process who represented multiple sectors of the US clinical trial enterprise.
Methods
The Expert Interviews Team nominated interview candidates considered as having extensive knowledge of and experience with informed consent in the United States and representing a diverse set of stakeholders. Invitation letters were sent to 42 candidates, and 25 agreed to participate.
Interviews were conducted by the Center for Information and Study on Clinical Research Participation (CISCRP) 9 between April and June 2014. The 1-h telephone interviews were conducted individually with each participant and followed a pre-specified interview guide designed to gather opinions on the current informed consent process, barriers to modification, and recommendations for actionable change. Transcribed interviews were analyzed for themes with the goal of identifying consensus opinions and recommendations. The project, including the interview guide, was approved by the Duke University Institutional Review Board (IRB), and each participant gave verbal informed consent prior to the interview (see Supplementary Appendix for detailed methods and a description of the interviewees).
Synthesis of results
Although all interviewees agreed that informed consent is essential to the protection of research participants, most also agreed that the informed consent process in the United States has evolved into a rigid and cumbersome process that requires serious attention and reform. From the interviewee recommendations, three major themes emerged with broad consensus: reform of the informed consent document, enhancing participant understanding of the clinical research, and modification of IRB review.
Informed consent document
The most frequent recommendation for a single actionable change was reform of the informed consent document that is required for federally regulated research—specifically, making it simpler, shorter, and more understandable. Interviewees observed that an unintended consequence of excessively long and complex informed consent documents is that research participants may not read or comprehend the information and simply sign the document. Half of the interviewees believed that the informed consent document was a deterrent to participation in clinical trials.
Some of the proposals could be implemented within the current regulatory framework, such as using modified formatting and graphics, ensuring that documents are written at the appropriate grade and health literacy level, and explaining technical terms in clear language. However, interviewees called attention to confusion around the extent of written information that must be provided to potential research participants under federal regulations. These regulations require that informed consent documents include eight mandatory elements (“basic elements of informed consent”) and six additional elements, if applicable (“additional elements of informed consent”). 10
Interviewees specifically raised concerns about excessively lengthy and detailed information related to the required disclosures of “the procedures to be followed in the study,” “any benefits to the subject or to others which may reasonably be expected from the research,” and “any reasonably foreseeable risks or discomforts to the subjects.” 10 Interviewees found the required description of “any reasonably foreseeable risks” to be particularly problematic. In recent draft guidance, the US Food and Drug Administration (FDA) recommended, “All possible risks do not need to be described in detail in the informed consent form, especially if it could be overwhelming for subjects to read. Information on risks that are more likely to occur and those that are serious should be included.” 11 Despite this regulatory guidance, the acceptable format and depth of how “any reasonably foreseeable risks” are presented to research participants may require future policy debate and potential modification of the federal regulations.
The importance of reforming the document to better inform research participants and increase participation in clinical research has been identified in other national policy forums, including those conducted by the Institute of Medicine.12,13 In the Advance Notice of Proposed Rulemaking cited above, the federal government also identified the improvement in informed consent documents, including addressing their excessive length and legalistic language, as one of eight major objectives. 6
Research participant understanding
The second most frequent recommendation from the interviews centered on creating a more interactive and ongoing process to enhance research participant understanding. The interviewees proposed multiple interventions that would not require national policy change and could be adapted to meet the needs of the specific study population (Figure 1). Examples included improving research staff training on the conduct of an informed consent discussion, conducting the process with adequate time and in a setting conducive to participant privacy and comfort, and providing an opportunity for prospective research participants to speak with other participants as a resource for information. The interviewees also emphasized the need for integration of the patient’s voice by including patients in the design of the informed consent process, including the informed consent document.
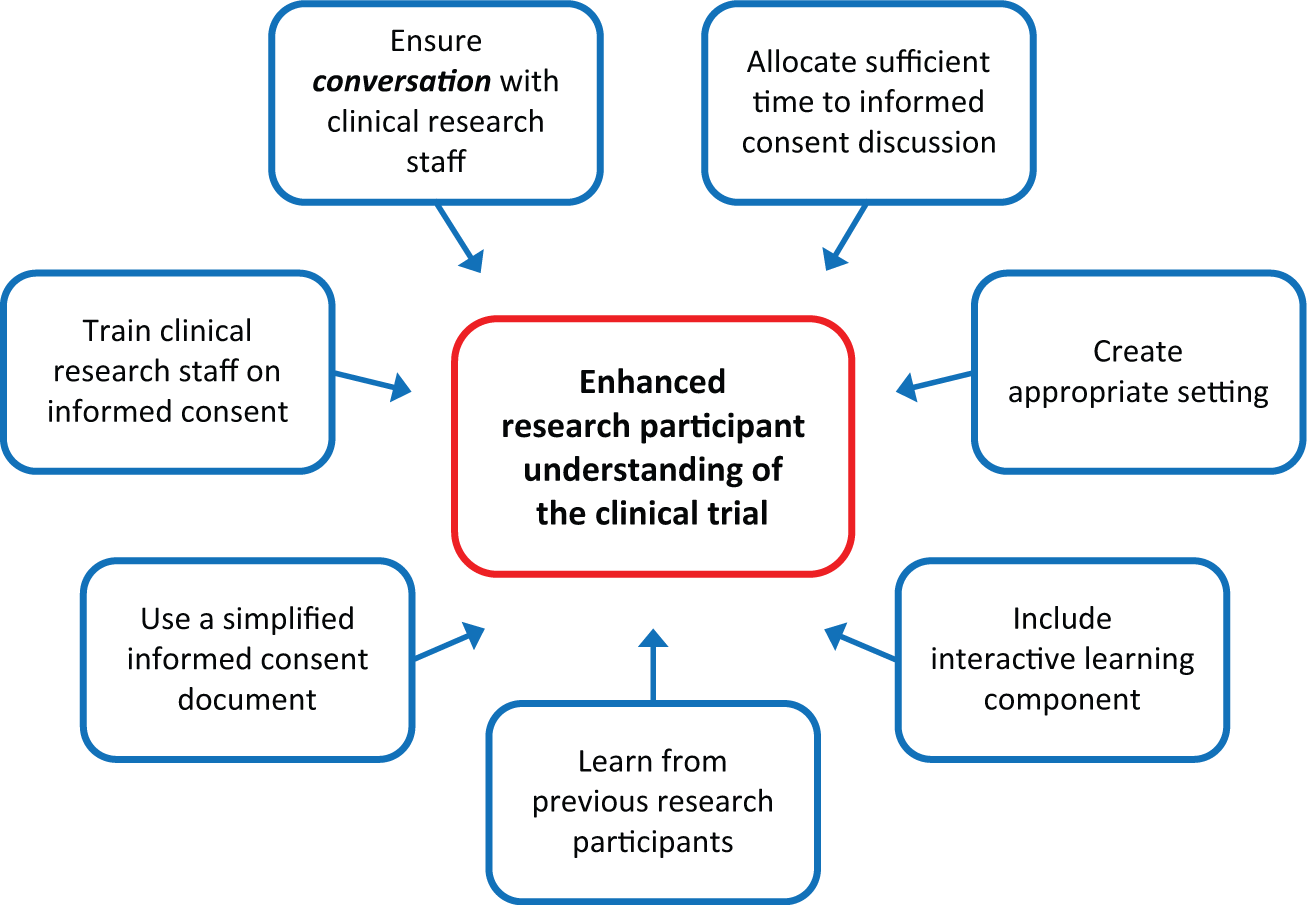
Enhancing research participant understanding of a clinical trial. Derived from authors’ analysis of results from 25 interview transcripts.
Evaluating research participant comprehension is another issue that arose during the interviews. There was consensus regarding a lack of evidence-based information about participant decision-making in clinical research. The interviewees also identified a lack of evidence-based tools and metrics to gauge participant comprehension specific to the informed consent process for clinical research. Among many scholars in this area, Brehaut et al. 14 conducted research that supports the notion that current informed consent documents and processes do not meet validated standards for promoting a decision-making process that allows “people to make explicit choices among clearly described options.” Several interviewees proposed that research participants should be tested to demonstrate a certain level of comprehension about the clinical investigation, but there was no consensus on this issue. Notably, although federal regulations generally require that a written consent document that embodies the required elements be signed by the participant or legally authorized representative, the regulations only require that “the investigator shall give either the subject or the representative adequate opportunity to read it before it is signed.” 15 Thus, current federal regulations do not require evidence of comprehension, and reform in this area is likely to be controversial.
IRB review
A third consensus issue from the interviews was the need for modification of practices in IRB review of clinical research. Some proposals likely could be implemented without national policy change. For example, several interviewees proposed that processes be implemented to help ensure that IRBs do not require the addition of language in informed consent documents that is intended to protect institutions from liability rather than enhance research participants’ understanding.
The most substantive recommendation was the transition to use a single IRB of record for multi-site clinical trials. A related CTTI project has recommended the use of a single IRB of record to improve the quality and efficiency of multi-center clinical trials and is working toward addressing barriers to adoption. 16 The National Institutes of Health recently developed a model of a single IRB of record for multi-site translational clinical research. 17 In the Advance Notice of Proposed Rulemaking, the Department of Health and Human Services observed that the current multiple IRB review system may “actually be leading to weaker protections for subjects” and proposed streamlining of IRB review of multi-site studies. 6 However, a federal statutory barrier exists for the use of a single IRB of record in FDA-regulated multi-site medical device clinical trials, unless a local IRB does not exist or its review is determined to be inadequate.6,18
Conclusion
While we recognize that modifying the informed consent process is a complex policy issue, our findings from interviews of a diverse group of experts not only solidify the recognition of major problems that require reform but also provide a framework for meaningful change at both the local and national levels. Several of the recommendations proposed by interviewees with broad consensus are feasible for rapid implementation within the current regulatory framework. However, certain proposed reforms will likely require change in federal regulations.
Footnotes
Acknowledgements
The authors thank Gina Uhlenbrauck, of Duke Clinical Research Institute, for editorial assistance in the preparation of this manuscript.
Declaration of conflicting interests
The authors have no potential conflicts of interest to disclose.
Funding
Funding for this project was made possible by the Food and Drug Administration through grant U19FD003800, as well as membership fees of CTTI participants. Views expressed in this publication do not necessarily reflect the official policies of the Department of Health and Human Services, nor does any mention of trade names, commercial practices, or organization imply endorsement by the United States Government. Views expressed reflect the opinions of the authors and do not necessarily reflect the opinions of their employers.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.