
Abstract
Background/Aims:
Several countries are working to adapt clinical trial regulations to align the approval process to the level of risk for trial participants. The optimal framework to categorize clinical trials according to risk remains unclear, however. Switzerland is the first European country to adopt a risk-based categorization procedure in January 2014. We assessed how accurately and consistently clinical trials are categorized using two different approaches: an approach using criteria set forth in the new law (concept) or an intuitive approach (ad hoc).
Methods:
This was a randomized controlled trial with a method-comparison study nested in each arm. We used clinical trial protocols from eight Swiss ethics committees approved between 2010 and 2011. Protocols were randomly assigned to be categorized in one of three risk categories using the concept or the ad hoc approach. Each protocol was independently categorized by the trial’s sponsor, a group of experts and the approving ethics committee. The primary outcome was the difference in categorization agreement between the expert group and sponsors across arms. Linear weighted kappa was used to quantify agreements, with the difference between kappas being the primary effect measure.
Results:
We included 142 of 231 protocols in the final analysis (concept = 78; ad hoc = 64). Raw agreement between the expert group and sponsors was 0.74 in the concept and 0.78 in the ad hoc arm. Chance-corrected agreement was higher in the ad hoc (kappa: 0.34 (95% confidence interval = 0.10–0.58)) than in the concept arm (0.27 (0.06–0.50)), but the difference was not significant (p = 0.67).
Limitations:
The main limitation was the large number of protocols excluded from the analysis mostly because they did not fit with the clinical trial definition of the new law.
Conclusion:
A structured risk categorization approach was not better than an ad hoc approach. Laws introducing risk-based approaches should provide guidelines, examples and templates to ensure correct application.
Introduction
In recent years, the increasing costs and administrative complexity of clinical research,1–5 as well as concerns about the shift of clinical trials from wealthy countries to less wealthy countries,6–8 have given rise to initiatives to revise the legal framework within which clinical research is conducted. The goal of these initiatives is to harmonize the clinical research processes, reduce the bureaucratic burden, lower the costs and better protect human beings participating in clinical trials.9–11 They recommend introducing so-called risk-based approaches to adapt regulatory requirements based on the risk associated with participation in clinical trials. Risk-based approaches have so far been used to guide on-site monitoring and quality assurance processes in the conduct of clinical trials.12,13 Nevertheless, the optimal framework for categorizing clinical trials according to their risks and the respective regulatory consequences remain unclear.
Two recent initiatives, one from the Organisation for Economic Co-operation and Development (OECD) working group and the other from a joint project of the Medical Research Council (MRC) and Medicines and Healthcare Products Regulatory Agency (MHRA), recommend introducing risk categories for clinical trials of investigational medicinal products in the regulatory framework.10,14,15 Furthermore, they propose adapting regulatory requirements for approval and conduct according to the risk category. Based on the marketing authorization status of the investigational medicinal product tested in the clinical trial, the OECD recommendations define three categories of clinical trials: A (lowest risk category), B and C (highest risk category). 14 The MHRA approach defines three types of clinical trials according to the risk associated with the investigational medicinal product compared with standard medical care: A (no higher), B (somewhat higher) and C (markedly higher). 15
The UK government has endorsed the concept of regulatory requirements proportionate to risk. 16 Furthermore, the European Union (EU) elaborated a new Clinical Trial Regulation 17 which introduces risk categories for clinical trials compatible with those recommended by the OECD. 14 Likewise, the United States is working to adapt their clinical trial regulations to align the complexity of the approval process with the risk to the participants.18,19 Switzerland became the first European country to adopt a risk-based legislation for clinical trials.20–22 A parliamentary motion mandated the Swiss Federal Council to introduce a federal act to harmonize pre-existing cantonal legislations on human research. 23 Consequently, a new article added to the Constitution provided the legal framework to regulate human research according to the risk to which participants are exposed. 24 To be compatible with other international initiatives, Switzerland broadly followed the OECD recommendations. 14 The draft Clinical Trials Ordinance which defines criteria to categorize clinical trials was released to public consultation between 31 August and 31 October 2012. 25 Online Appendix 1a compares the clinical trial categories defined in the draft Clinical Trials Ordinance with the EU regulation and those of the OECD and MHRA. In contrast to the EU regulation and the OECD and MHRA initiative, the draft Clinical Trials Ordinance regulates clinical trials involving any type of intervention and not only drugs or devices. Online Appendix 1b presents the risk categories for clinical trials with medical devices and non-pharmacological/non-device interventions. The new regulation does not establish a concept of minimal risk, and it does not consider ‘standard care’ as a principle for categorization. The draft Clinical Trials Ordinance categorization approach is based on two principles: (1) the marketing authorization status in Switzerland for drugs and medical devices and (2) whether the use of the investigated intervention falls within the specified summary of product characteristics (for drugs), Conformité Européenne (CE) mark specifications (for medical devices) or established medical guidelines (for non-pharmacological and non-device interventions). Drugs or devices with marketing authorization are assumed to have been tested and their use authorized by competent authorities so that potential risks related to the use of such medicinal products are reasonable for trial participants when used according to their (approved) specifications. Similarly, the risk of non-pharmacological/non-device interventions is assumed reasonable when used as indicated in an established medical guideline. Based on these principles, the sponsor has to classify the clinical trial into one of three categories (A, B or C). Each category implies different regulatory requirements for approval, conduct, insurance and safety reporting.20,26,27 Category A represents the category with the fewest and category C the one with the most regulatory requirements.
In this study, we tested the consistency and applicability of the criteria for categorization of clinical trials outlined in the draft Clinical Trials Ordinance. 25 For this purpose, we used a randomized controlled trial with a method-comparison study nested in each arm. We assessed how consistently clinical trials were categorized using the draft Clinical Trials Ordinance criteria (concept) compared to an intuitive, ad hoc approach without pre-specified criteria (ad hoc). We compared the categorization agreement between categorizing persons using the concept approach with the categorization agreement between categorizing persons using the ad hoc approach. Finally, we identified qualitative aspects related to the applicability of the categorization procedure and recommendations for implementation of similar legislations in the future. The results of this study served to inform policy-makers prior implementation of the Ordinance.
Methods
Study design
This was a two-arm randomized controlled trial with a method-comparison study nested in each arm. In one arm, different types of categorizing persons (assessors) used the risk-based categorization approach outlined in the new draft Clinical Trials Ordinance to categorize clinical trial protocols (concept). In the other arm, assessors used an unstructured, intuitive approach (ad hoc). The categories were the same in both arms; only the categorization procedure itself, that is, the guidance on how to arrive at the respective category, was different. We compared the categorization agreement among assessors between arms and within arms. We hypothesized that structured pre-defined criteria (concept) would lead to more consistent categorizations as compared to unstructured categorizations and consequently better agreement between assessors.
Concept categorization procedure
In the concept arm, assessors used a web-based, decision-tree questionnaire built with the categorization criteria defined in the draft Clinical Trials Ordinance. No specific training was provided besides a written general introduction. The criteria and number of applicable categories varied depending on three main aspects: first, the type of intervention studied, that is, drugs, medical devices or non-pharmacological/non-device intervention; second, the approval status in Switzerland for drugs and medical devices; and third, whether the intervention was used according to the summary of product characteristics in case of drugs, the CE mark specifications in case of medical devices or established medical guidelines in case of non-pharmacological/non-device interventions. Accordingly, assessors applied different categories: A, B or C for clinical trials with drugs; A or C for clinical trials with medical devices; and A or B for clinical trials with non-pharmacologic/non-device interventions. Category A represents the lowest and C the highest risk category. In case of clinical trials with mixed interventions (e.g. drug and device investigated in the same trial), each intervention was first categorized individually, and the highest risk category was assigned to the study as a whole. Online Appendix 2 presents the forms used to develop the web-based questionnaires for the concept arm.
Ad hoc categorization procedure
In the ad hoc arm, assessors did not have specific criteria to guide the categorization of clinical trial protocols. Assessors had sets of regulatory requirements of increasing strength for approval and conduct of clinical trials (e.g. compensation of damages, application procedure, dossier content and safety reporting) that correspond to each risk category. Assessors were asked to appraise the clinical trial protocol, to consider the potential risk to which participants would be exposed and to indicate the category of regulatory requirements that they considered most appropriate for approval and conduct. As with the concept arm, no specific training was provided besides a written general introduction. Assessors applied the same categories as in the concept arm: A, B or C for clinical trials with drugs; A or C for clinical trials with medical devices; and A or B for clinical trials with non-pharmacologic/non-device interventions. Online Appendix 3 lists the regulatory requirements by risk category and type of intervention.
Eligibility criteria of clinical trial protocols
Protocols were eligible if they had been approved by a Swiss ethics committee between 2010 and 2011 and described a clinical trial that fell within the definition introduced by the new law: ‘Clinical trial means: research project in which persons are prospectively assigned to a health-related intervention in order to investigate its effects on health or on the structure and function on the human body’, with health-related intervention defined as ‘a preventive, diagnostic, therapeutic, palliative or rehabilitative measure investigated in a clinical trial’. 20 No restriction was applied on the type of intervention that was evaluated or on the affiliation of the sponsor (academic or commercial).
Identification of clinical trial protocols
Of the 13 Swiss ethics committees, 9 were asked to participate in the study (Aargau, Bern, Basel, Lucerne, Geneva, Ticino, St. Gallen, Lausanne and Zurich). Ethics committees were asked to identify consecutive clinical trials and to seek consent to participate in the study from the corresponding sponsor. Ethics committees that received up to 100 submissions in 2011 (Aargau, Lucerne and Ticino) were asked to obtain consent from 20–30 sponsors. All other ethics committees (Zurich, Bern, Basel, Lausanne, Geneva and St. Gallen) were asked to obtain consent from 50 sponsors. Sponsors who agreed to participate were asked to provide an electronic copy of the latest approved version of the protocol or to allow the respective ethics committee to provide us with a copy.
Assessors of clinical trial protocols
There were three types of assessors categorizing protocols: a contact person (per trial) on behalf of the clinical trial sponsor (Sponsor), a member of each ethics committee (Ethics Committee) and an expert group (Expert Group). The Expert Group was made up of a clinical trial methodologist and physician from the clinical trials unit at the University of Bern (CTU Bern) along with three staff members of the Swiss Federal Office of Public Health who contributed to drafting the Clinical Trials Ordinance. All assessors were given the latest approved version of the protocol, and all based their categorization on the same version. Sponsors and corresponding clinical trial protocols were randomized to the concept categorization or the ad hoc categorization procedure arm. In a few cases, a sponsor was associated with more than one protocol, but the randomized categorization approach was similar across protocols (cluster randomization).
In the concept categorization arm, each clinical trial protocol was categorized by the corresponding contact person (Sponsor), a member of the ethics committee that had approved the clinical trial (Ethics Committee) and two members of the expert group (Expert Group). Each clinical trial protocol in the ad hoc categorization arm was categorized by the corresponding Sponsor and the Expert group. However, the Expert Group categorized all clinical trial protocols, including those in the ad hoc arm, using the concept categorization procedure. Composition of the Expert Group varied and was determined by availability (at least 2 out of 5 potential persons). Several assessment meetings were held and protocols assessed consecutively without any random mechanism. Agreement between members of the Expert Group was reached by consensus or by involvement of a third group member, if necessary.
Data collection
Data were collected via a web-based electronic data capturing system. Data collection processes were validated and tested prior to the study. Assessors were given a manual that explained the use of the system. Sponsors and Ethics Committees were sent up to five e-mail reminders during follow-up if they failed to respond.
Outcome measures
The primary outcome was the difference in categorization agreement between the Expert Group and the Sponsors across the arms as an indicator for consistency of categorizations. The secondary outcome measure was categorization agreement between assessors within the arms (inter-rater reliability).
Because of unexpectedly high disagreement between assessors about the category and type of intervention (see results), the Expert Group decided, post hoc, to re-assess 89 clinical protocols. Six weeks after the first assessment, the same persons who initially performed the first assessment performed a second assessment of 79 randomly sampled protocols where initial agreement was low and 10 randomly sampled protocols where initial agreement was high (intra-rater reliability).
Sample size
Sample size calculation was based on a simulation approach for the primary outcome and for the inter-rater reliability outcome. For the primary outcome, we determined sample size to compare chance-corrected agreements (Cohen’s kappa) between both arms. The study size should have given the study a power of 80%, with a two-sided type I error of 0.05 to detect a difference in kappas of 0.3 (0.8 in concept arm and 0.5 in ad hoc arm). The simulation resulted in a sample size of 210 trial protocols (105 pro arm). After taking the number of potential non-responders into consideration, we set the resulting minimal sample size to 120 protocols per study arm (240 overall).
To calculate the sample size for inter-rater reliability, we only considered the concept arm. The 95% confidence interval (CI) around the kappa coefficient of 0.8 should not be broader than ±0.2; that is, a kappa coefficient < 0.6 should be excluded. The simulation resulted in a sample size of 120 trial protocols. After considering potential non-responders, we set the definitive sample size for assessing inter-rater reliability at 140 trial protocols in the concept arm. The overall sample size for the study was therefore set to 260 trial protocols (140 for the concept and 120 for the ad hoc arm).
Randomization, concealment and blinding
Trial protocols were randomized in three batches to the concept or ad hoc arm, based on computer-generated random numbers. An independent data manager, who otherwise did not participate in the study, was responsible for the allocation. It was not possible to blind assessors.
Statistical analysis
Cohen’s linear weighted kappa was used to quantify agreement between categorizing bodies. Kappas and raw proportions of agreement were calculated using bootstrapping methods (N = 4000 repetitions), which allowed us to control for the fact that some sponsors evaluated multiple protocols. Kappa values from 0 to 0.4 were interpreted as low agreement; 0.40 to 0.75 were fair to good; and larger than 0.75 were excellent. For the primary outcome, we compared kappa values of rater-pairs by a z-test on the bootstrapped data. For stratified analyses, we compared subgroups by a z-test on the bootstrapped data (test for interaction).
These analyses were done with Stata 28 (StataCorp. 2011). Since Cohen’s kappa is an equivocal measure of reliability, prone to biases since it is dependent on the underlying distributions, we also computed Krippendorff’s alpha as an alternate measure of reliability. Since values of alpha and kappa were similar, we report only Cohen’s kappa, as specified in the original trial protocol. All CIs relate to the 95% level, and all reported p-values are two-sided.
Results
Eight of the nine anticipated Swiss ethics committees agreed to participate in the study (Aargau, Bern, Geneva, Lucerne, St. Gallen, Ticino, Lausanne and Zurich). We received 231 clinical trial protocols from 147 sponsors. Of these, 116 trial protocols were randomly allocated to the concept categorization procedure and 115 to the ad hoc procedure (Figure 1). Data collection lasted from 1 October 2012 to 16 January 2013. Sponsors categorized 102 protocols with the concept procedure and 103 with the ad hoc procedure (89%). Ethics Committees categorized 78 of 116 protocols allocated to the concept procedure. The Expert Group categorized 178 of 231 protocols. We included 142 trial protocols in the final analysis and excluded 89 (38.6%) for the following reasons: (1) studies did not meet clinical trial definition according to the legislation, (2) were duplicate studies or (3) other reasons (see Online Appendix 4 for details). Table 1 summarizes baseline characteristics of the trial protocols included in the analysis. Most sponsors were affiliated with academia (76%), and the most common therapeutic area of research was oncology (16%). Characteristics were balanced across the two randomized arms. Figure 2 illustrates categorization by the different assessors. The Sponsors categorized the majority of protocols into category A, while the Expert Group categorized the majority into category B. The Ethics Committee categorized most protocols either into category A or C. Online Appendix 5 presents the final categorization by type of assessor.
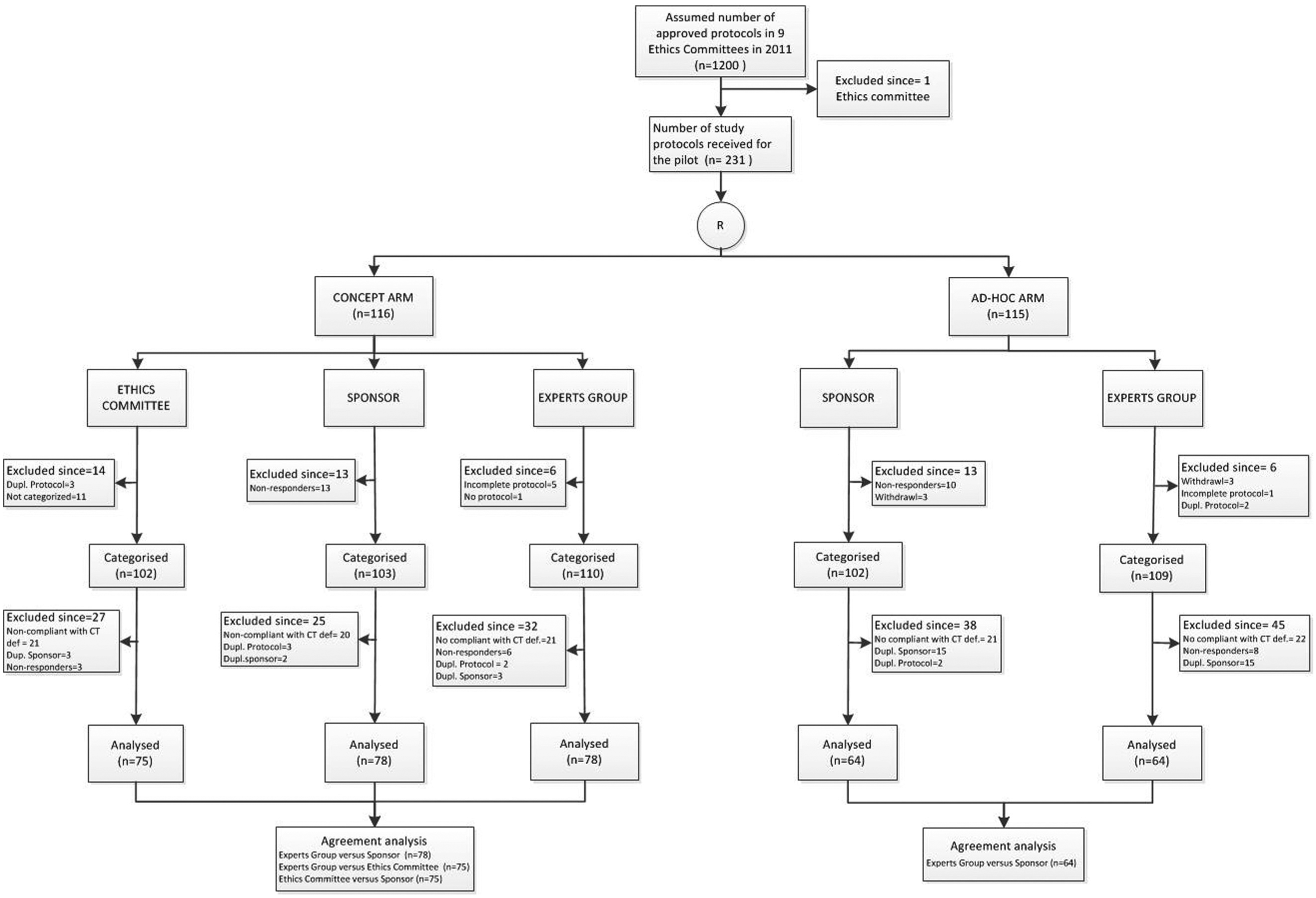
Flowchart of trial protocols categorized by arm and assessor.
Characteristics of clinical trials protocols.
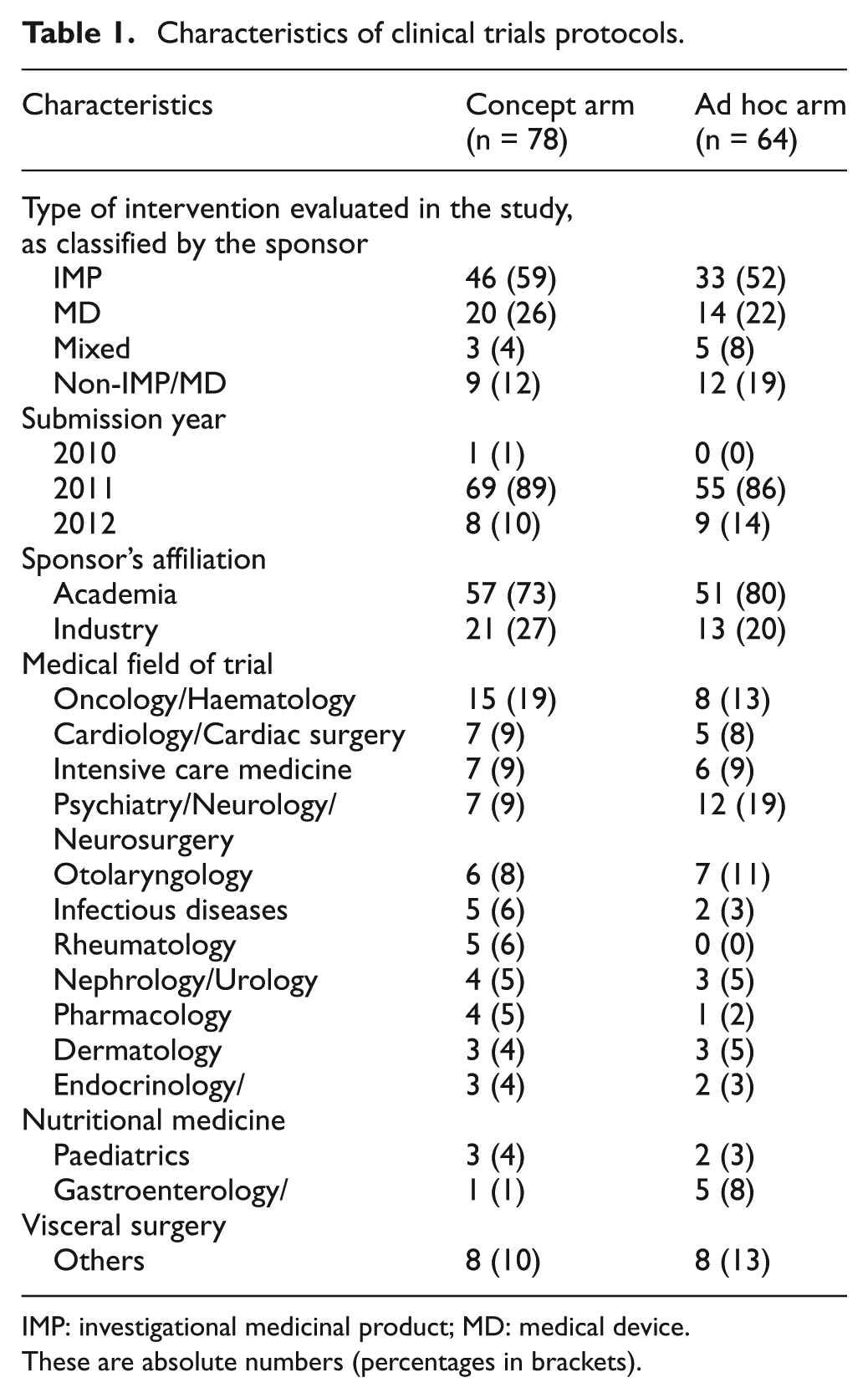
IMP: investigational medicinal product; MD: medical device.
These are absolute numbers (percentages in brackets).
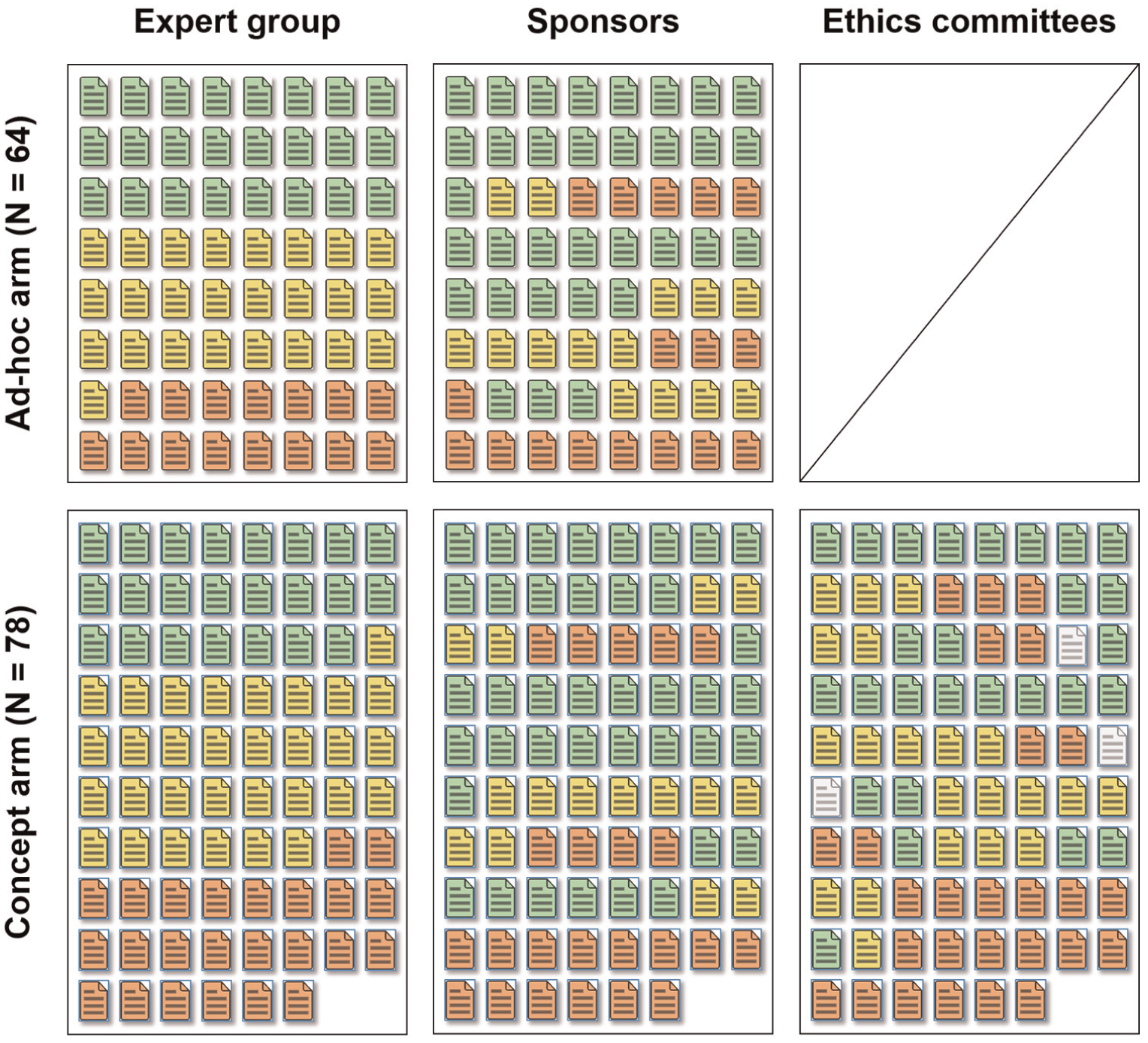
Assessments of individual protocols by arm and group. Each pictogram represents an individual protocol assessed in the trial across groups within each arm. Ethics committees did not assess protocols in the ad hoc arm.
Trial category: agreement between assessors within and between the concept and ad hoc arm
We compared agreement between assessors about the trial category in two ways: (1) within each arm, between the Sponsors and the Expert Group; and (2) between the arms. Raw agreement within the concept arm was 0.74 and within the ad hoc arm 0.78. There were major disagreements (e.g. if a trial was categorized by one body as A or C and by the other as C or A, respectively) for 13 of 78 protocols in the concept arm (17%) and for 8 of 64 protocols in the ad hoc arm (12%). Chance-corrected agreement between the Expert Group and Sponsors was low within the concept arm (kappa: 0.27; 95% CI = 0.06–0.50) and the ad hoc arm (0.34; 95% CI = 0.10–0.58). The difference between kappa values was 0.07 in favour of the ad hoc procedure (p-value: 0.67). Neither the type of sponsor, whether commercial or academic (p-value for interaction: 0.74), nor the type of intervention (p-value for interaction: 0.85) affected the degree of agreement between assessors (Figure 3).
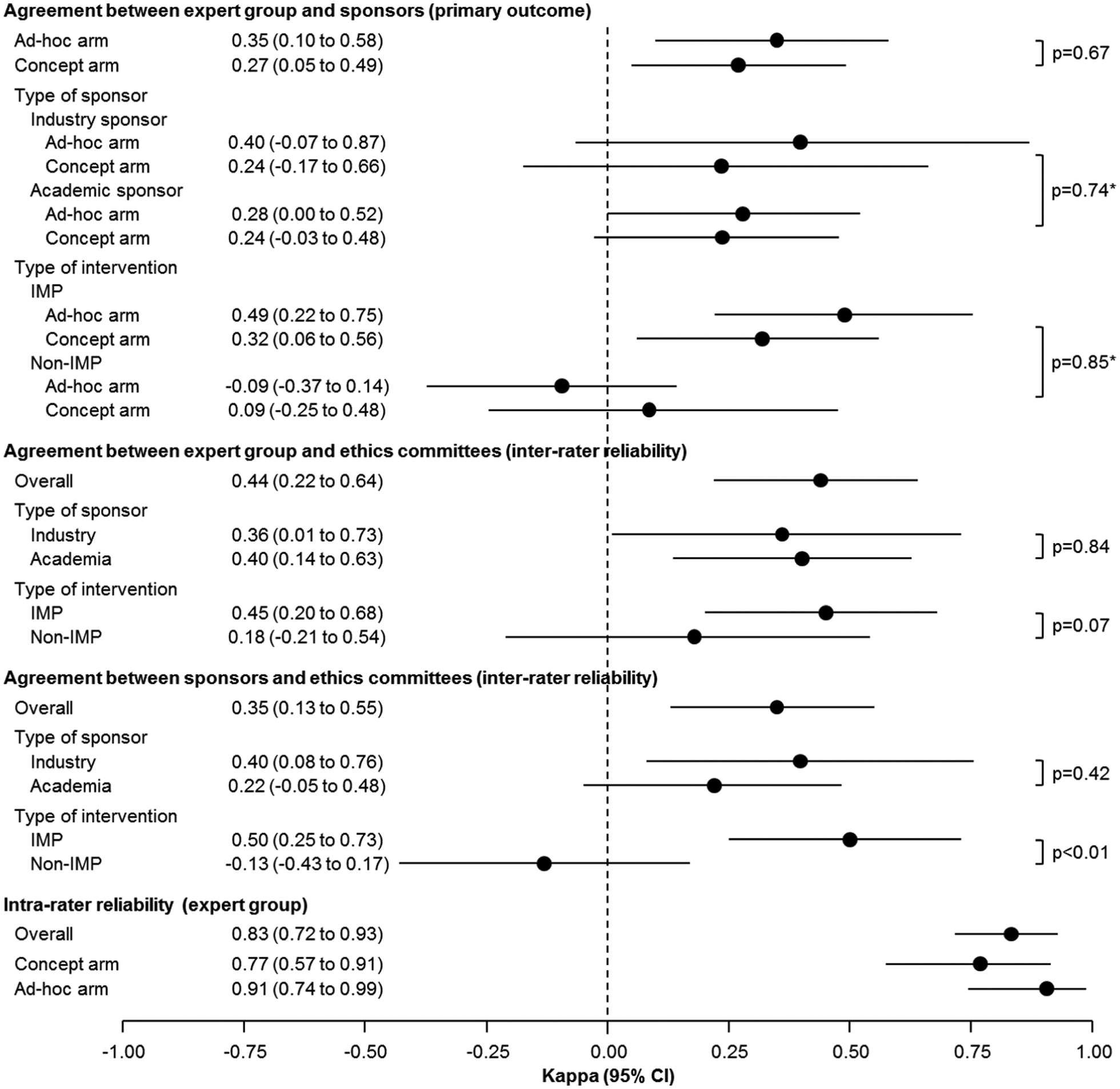
Caterpillar plot of primary and secondary outcomes.
Inter-rater reliability: agreement between assessors using the concept procedure
For the trial category, raw agreement between Ethics Committees and the Expert Group was 0.81 and between Ethics Committees and the Sponsor was 0.75. Chance-corrected agreement was low between Ethics Committees and the Expert Group (0.44; 95% CI = 0.22–0.64) and between Ethics Committees and the Sponsor (0.35; 95% CI = 0.13–0.55). Neither the intervention type nor the affiliation of the sponsor affected the level of agreement between assessors (Figure 3).
Type of intervention evaluated in the clinical trial – concept and ad hoc arm
The raw agreement on the type of intervention used in the trial was relatively high between assessors: Expert Group versus Sponsors was 0.82, Expert Group versus Ethics Committees was 0.83 and Sponsors versus Ethics Committees was 0.81. The chance-corrected agreement on the type of intervention evaluated in a given trial was generally moderate for the Expert Group versus Sponsors (kappa: 0.68; 95% CI = 0.53–0.84), Expert Group versus Ethics Committees (0.67; 95% CI = 0.52–0.82) and Ethics Committees versus Sponsors (0.65; 95% CI = 0.49–0.80).
Intra-rater reliability: agreement between first and second expert group’s categorization
Because of unexpectedly high disagreement between assessors about the category and type of intervention, the Expert Group decided, post hoc, to re-assess 89 clinical protocols. For the trial category, raw agreement between the categorization of the first and the second Expert Group was high (0.95). After adjusting for chance, the raw agreement was moderate (0.58; 95% CI = 0.30–0.80).
For the type of intervention evaluated in the trial, the agreement between the first and the second Expert Groups’ categorization was high. Raw agreement was 0.95, and the chance-corrected agreement kappa was 0.93 (95% CI = 0.84–0.98). Online Appendix 6 shows the agreement between the first and the second expert group categorization.
Discussion
In this study, we assessed how accurately and consistently clinical trials are categorized for regulatory purposes using two different approaches: an approach using criteria set forth in the new Swiss law on research in humans (concept) or an intuitive approach (ad hoc). We found no meaningful difference between the approaches. The concept procedure, when implemented without specific training, did not make categorization of trial protocols more consistent than the ad hoc approach. In the concept arm, major disagreements were present in more than 15% of protocols.
We expected that categorization of clinical trials would be straightforward with the presence of pre-defined criteria. For example, we assumed that trials investigating non-authorized drugs, falling into category C, would be easy to categorize, even for persons without specific training in the categorization procedure. However, this study showed that even well-structured categories for clinical trials are not consistently interpreted and that training is required to classify them. There was considerable disagreement between Sponsors, Ethics Committees and Experts about the category of clinical trials. Furthermore, we found disagreement between assessors about the type of interventions that were evaluated in the trial. For example, some assessors categorized homeopathic solutions and food supplements as drugs and others as non-drug chemical preparations.
Clinical trial protocols included in this study were written under the old law, which did not mandate an explicit delineation of intervention types for trials. Consequently, these protocols may be less clear than protocols written under the new law, which sets higher standards. Thus, we could expect fewer disagreements in real-world situations after the new law is implemented. Our data do not indicate that specific aspects of the categorization concept are in need of modification. Our subjective impression was that completeness of information required to categorize a trial was higher for industry protocols as compared to protocols from academia.
Limitations
This study was intended to inform policy-makers on the applicability of the categorization criteria proposed in the draft Clinical Trials Ordinance. Data collection started right after the public consultation. In the majority of cases, institutions and not individuals are invited to participate in public consultations. However, we do not know whether assessors randomized to the ad hoc procedure took part in the public consultation before our study commenced.
We excluded a large number of trial protocols (N = 89) from our final analysis. One of the reasons was that we could not contact Sponsors directly to ask them to participate in the evaluation. The participating Ethics Committees selected trial protocols that complied with the new clinical trial definition and then contacted the Sponsors to obtain their consent. We mostly excluded protocols selected by the Ethics Committees because they did not meet the new definition for clinical trials. After completion of this study and during feedback rounds, Ethics Committees acknowledged that they might not have fully understood the new definition at the time of selecting trial protocols for this study, and thus may have misclassified some studies. Since the new legislation was implemented without instructions or training sessions, misclassifications of type of research project may also happen in the future. Decisions to exclude studies were taken without knowledge of any categorization result of the sponsor or ethics committee. It is unlikely that these exclusions introduced bias, but they did reduce sample size and power.
To reduce the workload for Ethics Committees, we limited the required number of consenting sponsors to 20–50, based on the number of submissions they received per year. Although Ethics Committees were advised to select trial protocols consecutively, we were unable to monitor the selection process. We do not know how many Sponsors were contacted by the Ethics Committee before they obtained the required number of consenting Sponsors. We cannot exclude the possibility of selection bias, although we think it is unlikely that the sample of trial protocols is not representative of all trial protocols in Switzerland. Unfortunately, there was no central repository for clinical research projects approved by Ethics Committees in Switzerland, so we cannot evaluate representativeness.
This was in a sense a retrospective study since we tested the categorization procedure on trial protocols approved before the new law came into effect. This might have contributed to the observed discrepancies. In addition, the approval status of investigated drugs or devices might have changed between the date that the initial clinical trial was approved and the date we conducted our study. Sponsors might have categorized these trials based on the current approval status, while the Expert Group used the approval status and label information current at the time the clinical trial was initially approved. However, the number of such cases was low.
We did not embed a qualitative study to formally explore the subjective experience of participants (Sponsors and Ethics Committees) as they used the new categorization procedure. In retrospect, and given the unexpectedly negative results of this study, we feel that a nested qualitative study would have been useful. Unfortunately, we were constrained in time and financial resources and could not pursue a more extensive study. This was also the reason for not including ethics committees in the randomized phase of the study which would have helped to address our study question more comprehensively. Finally, the pre-specified sample size was not reached. Several of the preceding limitations contributed to this. Most important among these was the high number of protocols that had to be excluded and the pre-specified numbers of sponsors to be contacted by each ethics committee to reduce their workload. Nevertheless, our results even with this reduced sample size are unlikely to change substantially with an increased sample size.
Strengths of the study
To our knowledge, this is the first study to formally evaluate the application of a new risk-based categorization approach for clinical trials implemented at legislation level. Randomized controlled trials are optimal to evaluate the effects of interventions and are well suited for testing the risk-adapted categorization procedure of clinical trials proposed by the new Swiss legislation. Although we excluded a high number of protocols, our sample size was sufficient to exclude a relevant agreement with the new categorization procedure as implemented in this study. We actively engaged the main stakeholders in clinical research in Switzerland and raised their awareness for the upcoming changes introduced by the new legislation. We also provided an opportunity for sponsors, members of ethics committees, academic institutions, the pharmaceutical industry and government bodies to consider the details, implications and consequences of implementing the risk-adapted classification criteria and adapted regulations. As such, our study provided valuable new information about the effects of the new legislation to the concerned community.
Implications for further research
The concept of enacting risk-based regulations for human research is still new. More research is needed to illuminate aspects of this approach and to assess the extent to which risk-based regulations serve their purpose. We offer a feasible and useful approach to gathering reliable data about important aspects of drafted regulations before they are implemented. Comparable studies in other countries, with different legal systems, or in other fields of research should seek to overcome the limitations of our approach.
Implications for legislation on clinical research
The risk-adapted categorization procedure defined in the new Swiss law is based on recommendations from the OECD working group. 14 The categorization criteria are broadly comparable with the MRC/DH/MHRA concept applied in the United Kingdom 15 and that proposed in the current version of the revised EU regulation. 18 Therefore, the limitations encountered during this study might also be applicable to EU countries.
Conclusion and legislation recommendations
Categorization procedures based on strict criteria are not necessarily an improvement over intuitive categorization, especially when sponsors use instead informally an ad hoc approach based on comprehensive information on the resulting consequences of each category. Failure to properly implement the procedure may be due to lack of training. More guidance and directed training for sponsors and ethics committees might ensure that the categorization procedure of the ordinance is followed more strictly. In particular, we believe that developing a binding protocol template, thereby forcing sponsors to provide explicitly all relevant information, would improve the reliability of the categorization. We also suggest that agreement between sponsors and ethics committees be assessed periodically after the regulation is enacted.
Footnotes
Acknowledgements
We thank all ethics committees and sponsors for their contribution to this study. We also thank Peter Jüni for discussions and contributions on the study design and Muriel Helmers for data management support.
Declaration of conflicting interests
S.Z., A.C., B.E.M. and M.G. are employed by the Swiss Federal Office of Public Health (FOPH), the funder of the study. They were responsible for drafting the final version of the Ordinance that forms the basis of the categorization procedure evaluated herein.
Funding
This study was supported by the Swiss Federal Office of Public Health (FOPH).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.