
Abstract
Background
The prevalence of low testosterone levels in men increases with age, as does the prevalence of decreased mobility, sexual function, self-perceived vitality, cognitive abilities, bone mineral density, and glucose tolerance, and of increased anemia and coronary artery disease. Similar changes occur in men who have low serum testosterone concentrations due to known pituitary or testicular disease, and testosterone treatment improves the abnormalities. Prior studies of the effect of testosterone treatment in elderly men, however, have produced equivocal results.
Purpose
To describe a coordinated set of clinical trials designed to avoid the pitfalls of prior studies and to determine definitively whether testosterone treatment of elderly men with low testosterone is efficacious in improving symptoms and objective measures of age-associated conditions.
Methods
We present the scientific and clinical rationale for the decisions made in the design of this set of trials.
Results
We designed The Testosterone Trials as a coordinated set of seven trials to determine if testosterone treatment of elderly men with low serum testosterone concentrations and symptoms and objective evidence of impaired mobility and/or diminished libido and/or reduced vitality would be efficacious in improving mobility (Physical Function Trial), sexual function (Sexual Function Trial), fatigue (Vitality Trial), cognitive function (Cognitive Function Trial), hemoglobin (Anemia Trial), bone density (Bone Trial), and coronary artery plaque volume (Cardiovascular Trial). The scientific advantages of this coordination were common eligibility criteria, common approaches to treatment and monitoring, and the ability to pool safety data. The logistical advantages were a single steering committee, data coordinating center and data and safety monitoring board, the same clinical trial sites, and the possibility of men participating in multiple trials. The major consideration in participant selection was setting the eligibility criterion for serum testosterone low enough to ensure that the men were unequivocally testosterone deficient, but not so low as to preclude sufficient enrollment or eventual generalizability of the results. The major considerations in choosing primary outcomes for each trial were identifying those of the highest clinical importance and identifying the minimum clinically important differences between treatment arms for sample size estimation.
Potential limitations
Setting the serum testosterone concentration sufficiently low to ensure that most men would be unequivocally testosterone deficient, as well as many other entry criteria, resulted in screening approximately 30 men in person to randomize one participant.
Conclusion
Designing The Testosterone Trials as a coordinated set of seven trials afforded many important scientific and logistical advantages but required an intensive recruitment and screening effort.
Introduction
The prevalence of low testosterone levels in men increases with age, as does the prevalence of many symptoms and conditions that are similar to those that occur when men of any age develop testosterone deficiency due to known pituitary or testicular disease. These parallels suggest the possibility that the lower testosterone levels in elderly men contribute to many other age-associated conditions. Prior trials of the effects of testosterone treatment of elderly men, however, yielded equivocal results. The Testosterone Trials are a coordinated set of clinical trials designed to determine if testosterone treatment would benefit elderly men who have low testosterone concentrations and conditions of which low testosterone may be a cause or a contributor.
Cross-sectional studies, such as the European Male Aging Study [1], and longitudinal studies, such as the Baltimore Longitudinal Study of Aging [2], showed gradual decreases in testosterone with increasing age. As men age, they also experience decreased muscle mass [3,4] and mobility [5], decreased sexual function [6], decreased energy, anemia [7], decline in memory and other cognitive functions [8], decreased bone mineral density [9] and increased fractures [10], increased fat mass [11], and impaired glucose tolerance [12].
Several randomized, placebo-controlled trials studied the effects of testosterone treatment of elderly men with low–normal to slightly low serum testosterone concentrations. Testosterone treatment consistently increased lean mass and decreased fat mass [13–17] and tended to increase bone mineral density of the spine [18,19]. Effects on muscle strength and physical performance [13–15,17,20], sexual function [13,17,20], cognition [21,22], and energy [13,17,21] were inconsistent.
In 2003, a panel of the Institute of Medicine of the National Academy of Sciences concluded that the evidence did not demonstrate any clear beneficial effect of testosterone treatment of elderly men and recommended a coordinated set of clinical trials to determine if such treatment would have any benefit. It recommended not conducting a long-term trial to evaluate risk until efficacy had been demonstrated [23]. The Testosterone Trials were designed to implement this recommendation.
Methods
Choice of overall study design
The major feature of The Testosterone Trials design is a coordinated set of clinical trials to test the efficacy of testosterone treatment in elderly men who had a low serum testosterone and one or more clinical abnormalities that low testosterone may cause. Each trial was designed to test the effect of testosterone on a clinical condition that testosterone is known to improve in men who have severe hypogonadism due to known disease of the pituitary or testes. The rationale for coordinating many aspects of these trials – including the recruitment, screening, treatment, and monitoring for safety, as well as the study governance and management – was to facilitate uniform selection and treatment and to allow pooling of safety data.
Coordination also had two practical advantages. The first was cost-efficiency, since the same recruiting, screening, and study management was used for all trials. Costs also were reduced greatly because participants could, when they qualified, enroll in more than one trial, reducing the total number who had to be identified, screened, and treated. The second was simplifying study management by having a single steering committee, data coordinating center and Data and Safety Monitoring Board, and the same field sites for all trials.
Participant selection – inclusion criteria
The major factors considered in choosing inclusion and exclusion criteria for the common aspects of the trial (Table 1) were age, serum testosterone concentration, conditions that could interfere with the interpretation of the results, and conditions that testosterone treatment could exacerbate.
Common inclusion and exclusion criteria for The Testosterone Trials a

Abbreviated list. The complete list of the common inclusion and exclusion criteria is in the supplemental data section.
We chose 65 years as the lower age limit because it is a commonly used cutoff for studies of the elderly.
We used the serum total testosterone concentration for screening, even though it is the free testosterone that is biologically active, because assays for total testosterone are more accurate and better standardized than those for free, and usually, total testosterone accurately reflects the free [24]. In addition, total testosterone assays are more widely available than free testosterone assays and more commonly used in clinical practice.
We selected a serum testosterone value for inclusion intended to be low enough that it would be considered unequivocally low but high enough that we would meet recruitment targets that provided the desired statistical power. The lower limit of the normal range of total testosterone for young men usually is considered to be about 300 ng/dL early in the morning. Based on these considerations, we chose to include men whose serum testosterone was <250 ng/dL at 8 a.m. to 10 a.m. on two occasions; because this stringent requirement hampered recruiting, we later relaxed the criteria to <275 ng/dL on the first screen, <300 ng/dL on the second, and <275 ng/dL for the average of the two assays.
Participants also were required to have one or more symptoms that could be consequent to the low testosterone in order to qualify for one of the three main trials (see main trials, below).
Participant selection – exclusion criteria
A final consideration in participant selection was to exclude men who were at relatively high risk of having conditions that testosterone treatment may exacerbate, such as prostate cancer, benign prostatic hyperplasia, erythrocytosis, and sleep apnea (Table 1).
We excluded men who had a history of prostate cancer or prostatic intraepithelial neoplasia, men who had a palpable prostate nodule by digital rectal examination, and men who had a relatively high risk of undiagnosed prostate cancer by the Prostate Cancer Risk Calculator [25]. We adjusted the prostate-specific antigen (PSA) level to what it would have been had the participant’s serum testosterone been normal, using unpublished data from the European Male Aging Study (courtesy, Dr Frederick Wu) that showed a significant association between the serum PSA and testosterone concentrations in men ≥65 years whose serum testosterone was <460 ng/dL and PSA was <4 ng/mL. The regression coefficient was 0.00128 (p = 0.007), and the coefficient of determination (r 2 ) was 0.016. The correction factor ((460 − serum testosterone concentration) × 0.00128) was added to the measured PSA to give the PSA value used in the risk calculator.
We excluded men whose risk of any prostate cancer was >35% and risk of high-grade cancer was >7%, values we considered conservative. For example, a 75-year-old Caucasian man who had no family history of prostate cancer could have had a serum PSA no higher than 2.8 ng/mL to qualify by this criterion.
Benign prostatic hyperplasia is also dependent on testosterone and its conversion to dihydrotestosterone [26,27]. We therefore also excluded men who had symptoms of severe lower urinary tract symptoms, as defined by a score of >19 on the International Prostate Symptom Score (IPSS) questionnaire.
We excluded men who had hemoglobin concentrations >16.0 g/dL and men with diagnosed but untreated sleep apnea.
Because of a report of a large excess of cardiovascular events in another trial [28], we excluded men who had had a myocardial infarction or stroke within the previous 3 months or had systolic blood pressure > 160 mm Hg, or diastolic blood pressure > 100 mm Hg. Other exclusions are listed in Table 1.
Choice of trials and parameters
Prior evidence about which conditions of elderly men testosterone was most likely to ameliorate determined the choice of trials. The main trials were Physical Function, Sexual Function, and Vitality. Cognitive function testing was performed on all participants. The Anemia Trial included all participants in the three main trials who were mildly to moderately anemic at baseline. The Cardiovascular and Bone Trials were open to participants in the main trials who met additional specific entry criteria.
Treatment allocation and balancing
We allocated participants to receive testosterone or placebo based on the covariate-adaptive approach of minimization. Minimization allows balancing with a larger number of variables than stratified randomization [29,30]. Balancing variables included participation in each of the main trials, clinical site, mean screening testosterone concentration under or over 200 ng/dL, age under or over 75, current antidepressant use, and current phosphodiesterase-5 inhibitor use. We used the method of Taves [29] with the modification that treatment optimizing balance for each participant was assigned with 80% (rather than 100%) probability to maintain some randomness to the algorithm.
Testosterone treatment
The duration of testosterone treatment was chosen to be 1 year, in keeping with the Institute of Medicine recommendation to study efficacy in short-term trials [23].
For a testosterone preparation, we chose a gel applied to the skin, because these preparations increase the serum testosterone concentrations of most hypogonadal men to within the normal range, and most men find them easy to use. We chose AndroGel 1% in a pump bottle (AbbVie).
Testosterone and placebo kits were prepared by the central pharmacy and sent to sites with numbers incorporated into the data management system that linked participant identification number to a kit number when the participant was randomized.
The goal of testosterone treatment was to increase the serum testosterone concentration of the men assigned to the testosterone arm to within the normal range for young men and maintain it during the 1 year of treatment. The initial dose was 5 g of gel. The serum testosterone concentration was measured at months 1, 2, 3, 6, and 9 in a central laboratory (Quest Diagnostics Clinical Trials, Valencia, CA, USA). The dose of AndroGel was adjusted after each measurement, if necessary, to achieve that goal by an algorithm shown in Figure 1. The target range was initially 400–800 ng/dL, but was changed to 500–800 ng/dL after early trial data showed that the median testosterone concentration was 400–500 ng/dL, below the middle of the target range. Compliance was assessed by weighing used and unused pump bottles. To maintain blinding, when we changed the gel dose (number of depressions of the pump bottle) of a man in the testosterone arm, we simultaneously changed the dose of gel of a participant in the placebo arm who recently had had blood drawn at the same visit but at a different clinical site. Dose adjustment was prompted by messages from a designated Data Coordinating Center research coordinator to site investigators based on testosterone levels measured centrally and communicated to the designated Data Coordinating Center research coordinator.
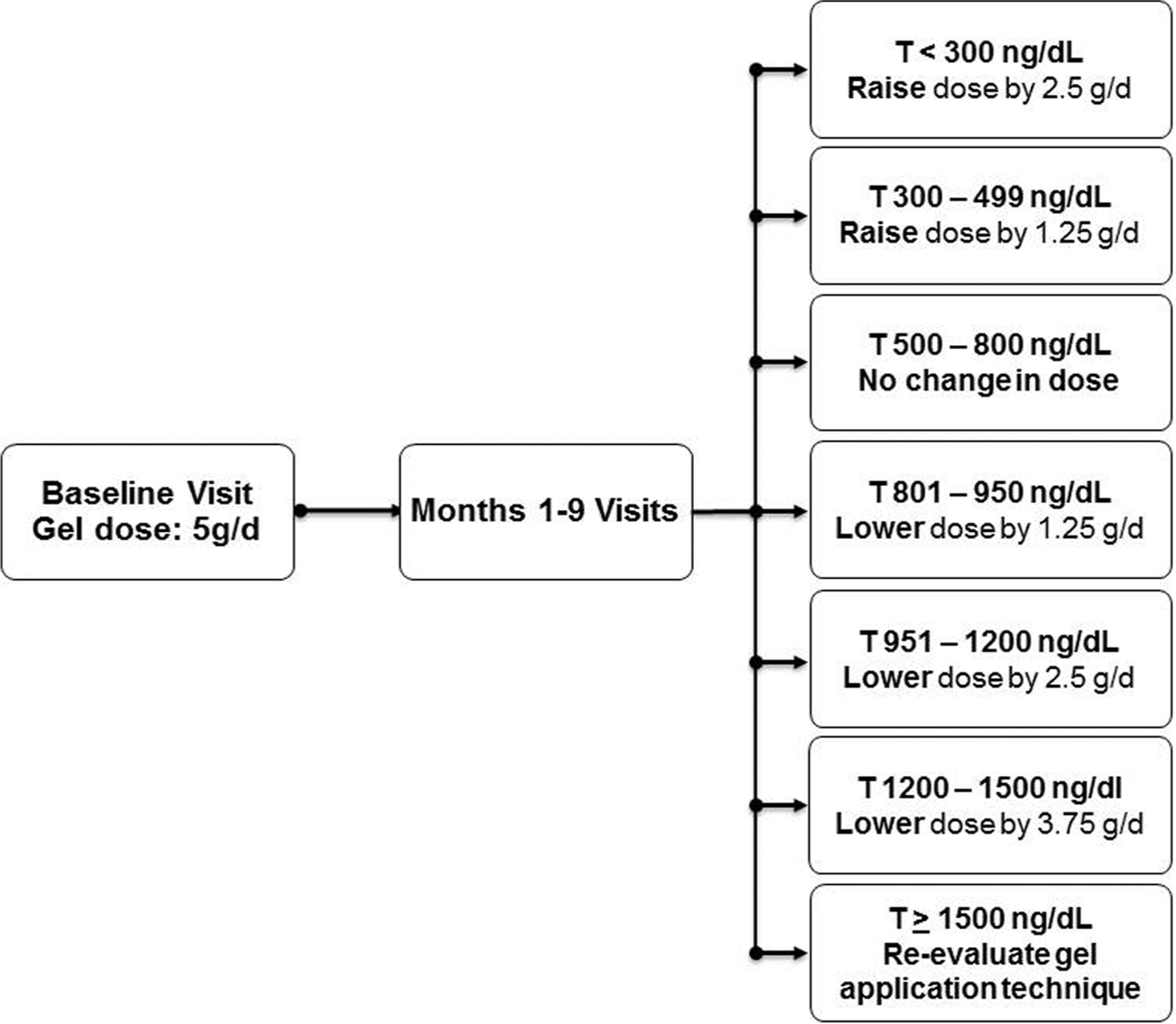
Algorithm for raising or lowering the dose of testosterone gel based on the serum testosterone concentration of a man in the testosterone treatment arm at months 1, 2, 3, 6, and 9.
We monitored the performance of the testosterone assay in collaboration with the Testosterone Standardization Program of the Centers for Disease Control by periodically inserting blinded aliquots of testosterone pools into the assay.
Monitoring participants for potential adverse effects of testosterone
Participants were monitored during the 1 year of treatment for development of conditions that testosterone could exacerbate. Participants were monitored for prostate cancer by digital rectal examination and serum PSA at 3 and 12 months. They were referred to a study urologist for consideration of a prostate biopsy whenever a nodule was detected or the PSA concentration increased by >1.0 ng/mL above the baseline value and was confirmed by a second measurement. If a biopsy showed prostate cancer, gel treatment was discontinued.
Participants were monitored for severe lower urinary tract symptoms by administration of the IPSS at months 3 and 12. An increase to >19 resulted in a review of possible causes. If none was found, treatment with an alpha-adrenergic receptor blocking drug was considered. If symptoms persisted, the participant was referred to a urologist for evaluation.
Participants were monitored for development of erythrocytosis by assaying the hemoglobin concentration at months 3, 6, 9, and 12. Whenever the value increased to >17.5 g/dL, confirmed by a repeat test, a cause was sought. If none was found, the gel dose was reduced. If the value did not decrease to ≤17.5 g/dL, phlebotomy was recommended.
Specific trials
To qualify for The Testosterone Trials overall, a man had to meet all of the general enrollment criteria described above (Table 1) and also the specific eligibility criteria for at least one of the three main trials (Table 2). A participant who also met the specific enrollment criteria for the Cardiovascular Trial and the Bone Trial could enroll in them (Table 2). Each specific trial had its own hypotheses, primary and secondary endpoints (Table 3), and sample size targets (Table 4).
The Testosterone Trials: additional inclusion and exclusion criteria for specific trials a

DISF-M-II SR: Derogatis Inventory of Sexual Functioning–Males–II–Revised; FACIT: Functional Assessment of Chronic Illness Therapy; DXA: Dual Energy X-Ray Absorptiometry.
Abbreviated list. The complete list of inclusion and exclusion criteria for specific trials is in the supplemental data section.
One of the main trials.
Tests of efficacy in The Testosterone Trials
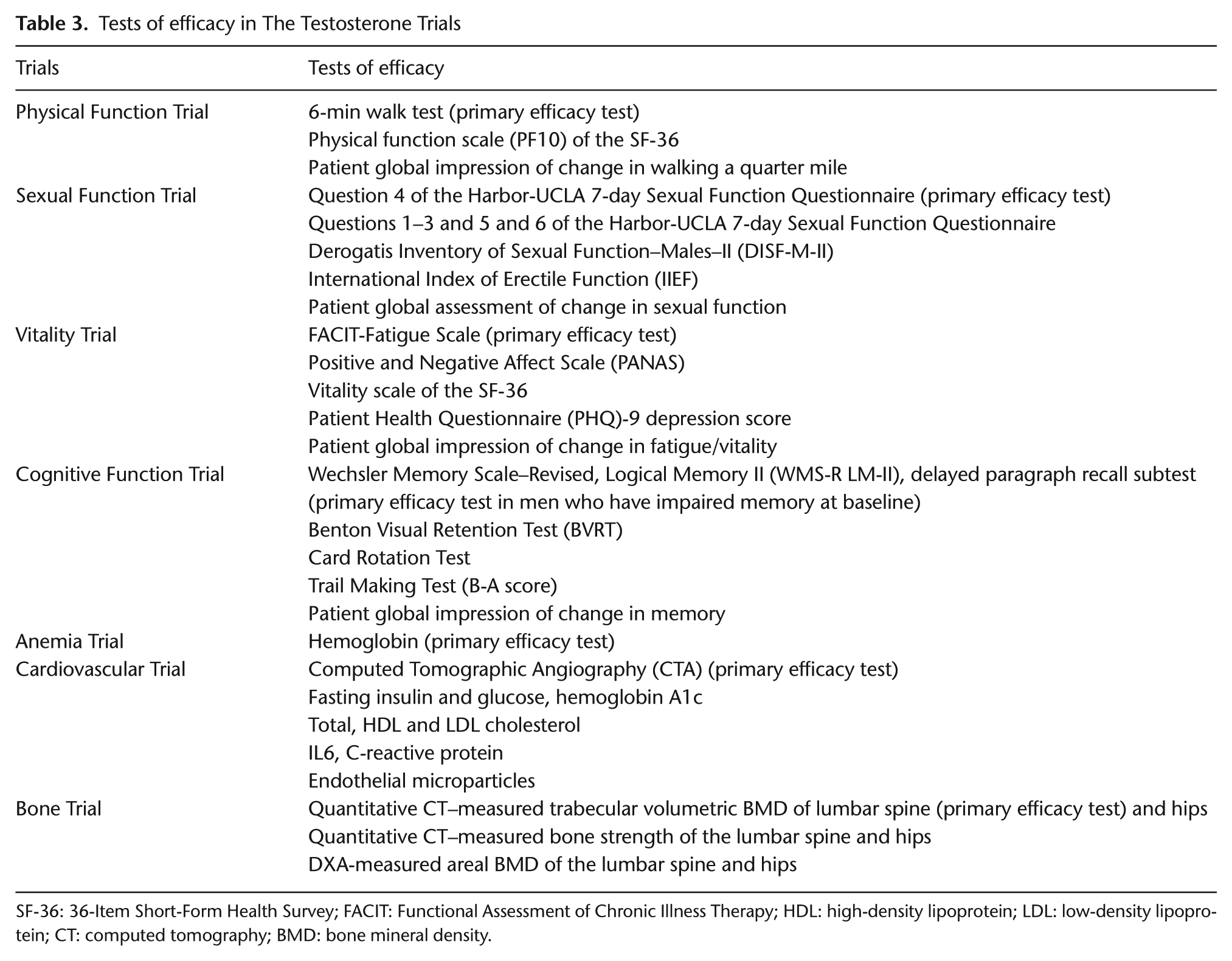
SF-36: 36-Item Short-Form Health Survey; FACIT: Functional Assessment of Chronic Illness Therapy; HDL: high-density lipoprotein; LDL: low-density lipoprotein; CT: computed tomography; BMD: bone mineral density.
Minimum detectable treatment differences for primary outcomes and targeted sample sizes per arm for each trial
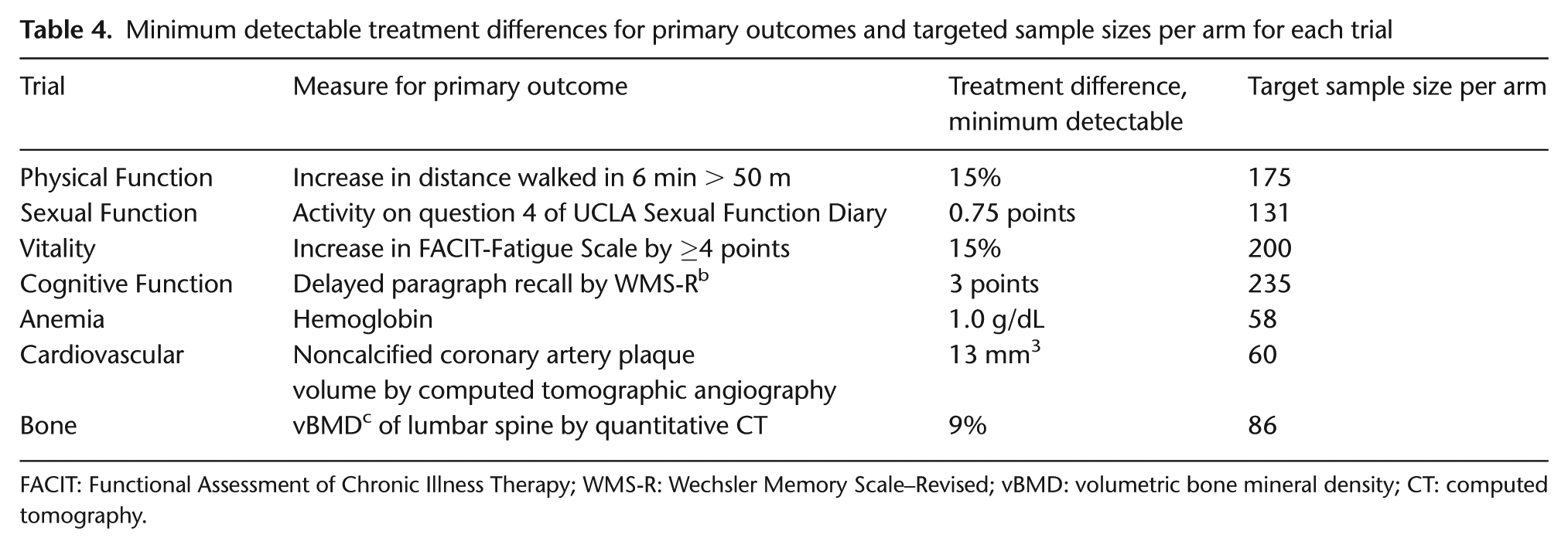
FACIT: Functional Assessment of Chronic Illness Therapy; WMS-R: Wechsler Memory Scale–Revised; vBMD: volumetric bone mineral density; CT: computed tomography.
Physical function trial
The primary hypothesis of the Physical Function Trial was that 1 year of testosterone treatment of elderly men with a low serum testosterone concentration and mobility disability would increase the proportion of men who improved their distance in the 6-min walk test by ≥ 50 m. This test was selected as the primary outcome because (1) walking is essential for most activities of daily living and predicts meaningful clinical outcomes [31–33] and (2) data about the minimum clinically important difference (50 m) were available [34–36]. One investigator (T.W.S.) trained the staff at each site in the administration of this test and visited each site once a year to monitor performance. The sample size estimate was based on providing 90% power to detect a meaningful difference if ≤15% of men in the placebo group and ≥30% of men in the testosterone group increased the distance they walked in 6 min by ≥50 m.
The inclusion criterion for the Physical Function Trial was symptomatic and objective mobility disability, defined by a combination of self-reported difficulty walking one-quarter mile and/or walking up one flight of stairs and a gait speed of <1.2 m/s on the 6-min walk test, a value associated with reduced survival [37,38].
Sexual function trial
The primary hypothesis of the Sexual Function Trial was that testosterone treatment of elderly men with low sexual interest will increase sexual activity, as assessed by question 4 of the Harbor-UCLA 7-day Sexual Function Diary [39]. This questionnaire had been used in previous testosterone trials to demonstrate increased sexual activity [40–42]. We administered it by interactive voice response [43]. The sample size estimate was based on providing 90% power to detect a difference in score of 0.75 units (standard deviation (SD) of change from baseline to 12 months of 1.86) [41].
The inclusion criterion was decreased libido, both self-reported and by a score of ≤20 on the Derogatis Inventory of Sexual Function–Males–II questionnaire, and a partner willing to have sexual intercourse at least twice a month.
Vitality trial
The primary hypothesis of the Vitality Trial was that testosterone treatment of elderly men with low vitality would increase the proportion whose score on the Functional Assessment of Chronic Illness Therapy (FACIT)–Fatigue Scale would increase by ≥4 [44]. The sample size estimate was based on providing 90% power to detect a difference of 15% (20% of men in the placebo group and 35% of men in the testosterone group) in the proportion of men showing would exhibit an increase at least this great. The inclusion criterion for the Vitality Trial was low vitality, as defined by both self-reported decreased energy and a score of <40 on the FACIT-Fatigue Scale.
Cognitive function trial
The primary hypothesis was that testosterone treatment of elderly men with age-associated memory impairment would result in greater improvement, or less decline, in verbal memory, as assessed by the Delayed Paragraph Recall Subscale (Logical Memory II) of the Wechsler Memory Scale–Revised [45]. Men considered to have age-associated memory impairment had subjective memory complaints, determined by their score on the Memory Assessments Clinics Questionnaire, and objective memory impairment, as determined either by a score on the Delayed Paragraph Recall Subscale or by Benton Visual Retention Test > 1 SD below the performance for young men. We calculated that a sample size of 235 men/arm would provide 90% power to detect a difference between the two arms of 3 points in the Wechsler memory scale, the difference between the 50th percentile for 70- to 75-year-old men (17) and that of 45- to 54-year-old men (20). Based on studies showing that about 60% of men over 65 years meet the criteria for age-associated memory impairment, and a total enrollment of 789, we expect this number of men with age-associated memory impairment.
The Wake Forest Cognitive Function Coordinating Center trained, certified, and recertified semiannually the site personnel who administered the tests.
Anemia trial
The primary aim was to determine if testosterone treatment would increase hemoglobin by 1.0 g/dL in men with unexplained mild to moderate anemia of the elderly, defined as a hemoglobin concentration 10.0–12.7 g/dL not due to nutrient deficiency, renal insufficiency, inflammation, or known hematologic disease. We expect to enroll about 100 men who met this criterion, which would provide >90% power to detect this difference.
Cardiovascular trial
The primary hypothesis was that testosterone treatment would decrease progression of noncalcified coronary artery plaque volume, as assessed by computed tomography (CT) angiography. Data using similar CT angiography techniques suggested that scans at baseline and 12 months for 60 men/arm would provide 80% power to detect a 13 mm 3 or larger difference between treatment arms after 1 year of treatment, a value chosen to be more conservative than the 14 mm 3 treatment effect of statins (Dr Budoff, unpublished). Dr Budoff trained the site technicians who performed the CT angiography.
Bone trial
The primary hypothesis was that testosterone treatment for 1 year would increase trabecular volumetric bone mineral density of the lumbar spine, measured by quantitative CT. A prior study showed that testosterone increased volumetric bone mineral density by 9% (standard error 3%) in 1 year [46]. With 86 men per arm, we should have 90% power to detect a difference this large or larger.
The quantitative CT Reading Center (ON Diagnostics) trained the site CT technicians who performed the quantitative CT scans and monitored scan quality. The University of California San Francisco Dual Energy X-ray Absorptiometry Quality Assurance Group trained the trial site dual energy X-ray technicians and monitored scan quality.
Recruitment and screening
Several methods of recruitment were used initially, including mass mailings and advertisements in newspapers and on radio and television. Mass mailings, based on lists specific for gender, age, and zip code, were most successful.
Screening was conducted in three successive steps (Figure 2, Tables 5 and 6). Men who responded to the advertisements were prescreened by telephone to exclude those who had no qualifying symptom or who had a disqualifying disease history. Potentially eligible men were asked to come to the trial site for the first in-person screening visit.
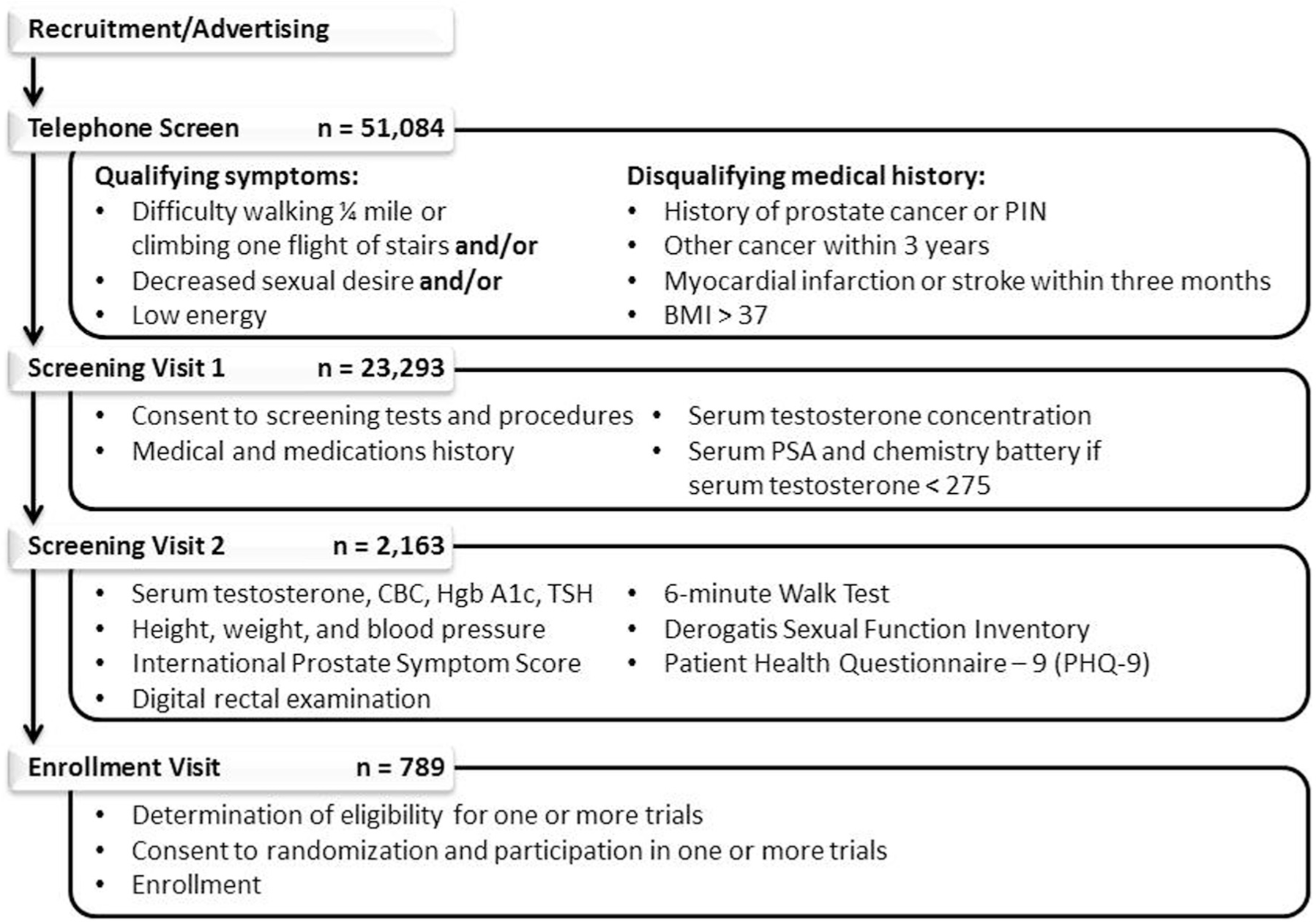
The stepwise screening process of The Testosterone Trials from the telephone screen to enrollment.
Testosterone trial test schedule
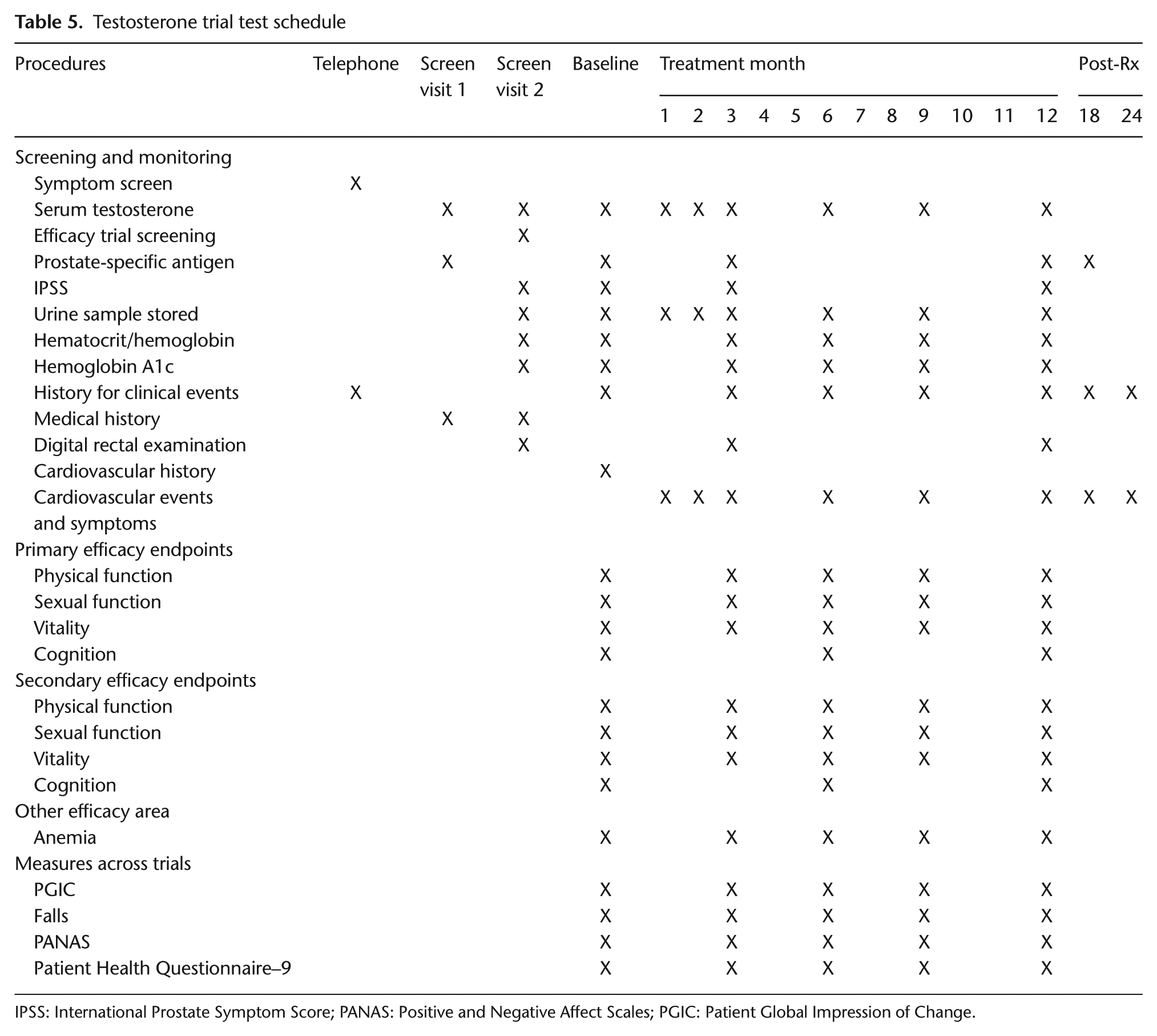
IPSS: International Prostate Symptom Score; PANAS: Positive and Negative Affect Scales; PGIC: Patient Global Impression of Change.
Targeted and actual enrollment in the seven testosterone trials

Actual enrollment in the main trials was greater than targeted because they were kept open to enable the Cardiovascular and Bone Trials, which did not begin enrolling until 14 months and 18 months later than the main trials, to complete enrollment.
Cognitive function testing was performed on all participants in the Physical Function, Sexual Function, and Vitality Trials.
All participants who were anemic were enrolled in the Anemia Trial.
At the first in-person screening visit, potential participants had a more detailed medical history and had blood drawn for serum testosterone concentration. Whenever the value was <275 ng/dL, a PSA and comprehensive metabolic panel were measured on saved serum. As long as the risk of prostate cancer, as determined by the Prostate Cancer Risk Calculator, was sufficiently low, the potential participant was asked to return for a second in-person screening visit.
At the second in-person screening visit, a second measurement of testosterone, a digital rectal examination, and other tests were performed to determine if the candidate participant qualified for any of the three main trials. Potential participants who qualified for at least one main trial on the basis of these tests were asked to schedule a baseline visit.
Adverse events
Although there are seven trials for efficacy, adverse events are collected and treated as if all participants are enrolled in one study.
Statistical considerations
Treatment blinding
Several methods were used to maintain blinding. Only the Data Coordinating Center and the Central Pharmacy knew treatment assignment. The testosterone and placebo preparations looked, smelled, and felt the same. Clinical sites did not have access to laboratory tests, such as testosterone, PSA, and hemoglobin concentrations, that testosterone treatment might increase.
Sample size considerations
Although we collected primary endpoint data in the three main trials at baseline, 3, 6, 9, and 12 months and will use longitudinal methods to analyze outcomes, we calculated sample sizes based only on comparisons between baseline and 12-month values. We inflated each sample size by 5% to compensate for participants with no post-baseline measures. For the Bone and Cardiovascular Trials, which had only one post-baseline measure, the sample sizes were inflated by 15% to account for dropouts. The Cardiovascular Trial was inflated by an additional 10% to account for a decrease in renal function after baseline and resulting ineligibility to receive contrast material at the 12-month visit.
Analytic methods
Each of the trials is considered a separate trial, so we shall analyze the efficacy results of each trial individually. The primary and secondary outcomes of each trial will be evaluated for those participants who enrolled in that trial. Participants will be analyzed in the group to which they were assigned, regardless of compliance with treatment. Each of the seven trials will be analyzed according to a pre-specified analytic plan.
Primary analyses of outcomes from all time points will be performed with random effects models for longitudinal data. Logistic models will be used for binary variables and linear models for continuous variables. Outcomes with measures at baseline and 12 months only (Cardiovascular and Bone Trials) will be compared using multivariate logistic regression for binary variables and multivariate linear regression for continuous variables, with adjustment for balancing factors at baseline. Dichotomous outcomes will be used for analysis when there are known minimal clinically important differences, to determine if testosterone not only has a statistically significant effect but also one of clinical significance. The potential impact of missing data on the trials’ outcomes will be assessed by sensitivity analyses. The independent Data and Safety Monitoring Board reviews data twice each year.
Results
Status of trials
Enrollment began in June 2010 and finished in June 2013. Of the 788 participants, 98 enrolled in all three main trials, 348 enrolled in two main trials, and 341 enrolled in only one main trial. Numbers of men enrolled in each trial are given in Table 6. We expect treatment will be completed by June 2014.
Lessons learned
Initial recruitment techniques included mass mailings, radio and television advertisements, and presentations about the trials to physicians and seniors. After a year, we found that most enrollees came from mass mailings, so subsequently, we employed this method primarily.
The screening process was conducted in three steps (Figure 2). Of approximately 64 men who expressed interest, only one enrolled. This attrition was the result of many entry criteria, especially the serum testosterone criterion (see above). By the initial criterion, only 7.8% of screenees qualified. By the relaxed criterion, 14.0% qualified.
Discussion
Many factors influenced the design of The Testosterone Trials. When selecting participants, the most important inclusion criterion was the serum testosterone concentration, because the goal of the study was to determine if raising the testosterone levels of elderly men with unequivocally low testosterone would benefit them. In calculating the required sample size for each of the seven trials, the most important decision was to set the power at 90% for all three of the main trials and for three of four remaining trials so that both positive and negative outcomes would be assessed with a high level of precision and thus have important implications for clinical practice. In choosing specific outcomes, we aimed for those that were clinically relevant. We also chose differences between treatment arms that would be clinically meaningful.
Perhaps the most unusual aspect of the study design was conducting seven trials, each with its own outcomes, under the umbrella of a single study. This structure had several advantages. The major scientific advantage was standardization of common entry criteria (e.g., testosterone concentration), treatment, and monitoring. Because of this standardization, findings from the seven trials can be compared with each other more readily. The logistical and financial advantages of this design were having one governing structure, one data coordinating center, one group of clinical trial sites, and one set of supporting teams. There was an even larger financial advantage by enrolling participants in more than one trial whenever eligible, since approximately half of the study cost was for recruitment.
Allowing participants to enroll in more than one trial also had a disadvantage. Although qualified participants could participate in all three main trials, as well as the Cardiovascular and Bone Trials, the number of tests that could be done in any one trial had to be limited to reduce participant burden.
A limitation of these trials is that the total number of subjects (789) is not nearly large enough, nor the duration of observation (2 years) long enough, to determine if testosterone treatment increases the risk of conditions that may be testosterone-dependent. We estimated that it would take 6000 men randomized to testosterone or placebo and followed for 6 years to determine if testosterone increases the risk of prostate cancer by 30%.
The Testosterone Trials, in summary, are a highly coordinated group of seven randomized, placebo-controlled, multicenter trials designed to determine if testosterone treatment will benefit elderly men with low serum testosterone concentrations and conditions for which low testosterone may be a cause or a contributor. The advantages of the study design included common entry criteria, treatment, and monitoring; the efficiency of having the same steering committee, data coordinating center, Data and Safety Monitoring Board, and trial sites; and the efficiency of participant enrollment in more than one trial. If these trials demonstrate sufficient benefits, a larger trial of longer duration will be necessary to assess safety.
Footnotes
Appendix
Acknowledgements
We thank many individuals who provided data prior to publication that aided greatly in the design of The Testosterone Trials, including Drs Frederick Wu, David Lee, Eric Orwoll, Mary McDermott, Donald Brambilla, John McKinley, Donna Ankerst, and Kirk Esley. Many other individuals gave invaluable advice in the early stages of the design of the study, including Drs Michael Barry, H. Ballantine Carter, Monique Cherrier, Suzanne Craft, Leonard Derogatis, Jerri Janowsky, Ira Katz, Scott Moffatt, and Ian Thompson. We thank Dr Renee Moore and Yawei Zhang, Shannon Chuai, and Liyi Cen for their support in developing the statistical aspects of the study design, and Dr Kenneth Rockwell for designing the drug labeling and distribution processes. We thank Dr Alisa Stephens for critical suggestions in drafting this article. We thank Sandra Smith for excellent assistance with preparation of the manuscript.
Funding
The Testosterone Trials were supported by a grant from the National Institute on Aging, National Institutes of Health (U01 AG030644), supplemented by funds from the National Heart, Lung and Blood Institute, National Institute of Neurological Diseases and Stroke, and National Institute of Child Health and Human Development. The Bone Trial was supported by a grant from the National Institute on Aging (R01 AG037679). The Anemia Trial was supported by a grant from the National Institute on Aging, National Institutes of Health (U01 AG034661) to the Partnership for Anemia Clinical and Translational Trials in the Elderly consortium. AbbVie (formerly Solvay and Abbott Laboratories) generously provided funding, AndroGel, and placebo gel. A.M.M. was supported by the Department of Veterans Affairs Puget Sound Health Care System. T.M.G. was the recipient of a Midcareer Investigator Award in Patient-Oriented Research (K24-AG021507) and is the recipient of an Academic Leadership Award (K07AG043587), both from the National Institute on Aging. The Yale Field Center was partially supported by the Claude D. Pepper Older Americans Independence Center (P30-AG021342). S.M.R. was supported by the Intramural Research Program, National Institute on Aging, National Institutes of Health. C.E.L. was supported by the National Institute for Diabetes, Digestive and Kidney Diseases, National Institutes of Health (DK079626) to the UAB Diabetes Research and Training Center. J.A.C. was supported by the National Institute on Aging, National Institutes of Health (R01 AG37679).
Conflict of interest
For a full declaration, please see supplementary data online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.