
Abstract
Background
Randomized clinical trials, particularly for comparative effectiveness research (CER), are frequently criticized for being overly restrictive or untimely for health-care decision making.
Purpose
Our prospectively designed REsearch in ADAptive methods for Pragmatic Trials (RE-ADAPT) study is a ‘proof of concept’ to stimulate investment in Bayesian adaptive designs for future CER trials.
Methods
We will assess whether Bayesian adaptive designs offer potential efficiencies in CER by simulating a re-execution of the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) study using actual data from ALLHAT.
Results
We prospectively define seven alternate designs consisting of various combinations of arm dropping, adaptive randomization, and early stopping and describe how these designs will be compared to the original ALLHAT design. We identify the one particular design that would have been executed, which incorporates early stopping and information-based adaptive randomization.
Limitations
While the simulation realistically emulates patient enrollment, interim analyses, and adaptive changes to design, it cannot incorporate key features like the involvement of data monitoring committee in making decisions about adaptive changes.
Conclusion
This article describes our analytic approach for RE-ADAPT. The next stage of the project is to conduct the re-execution analyses using the seven prespecified designs and the original ALLHAT data.
Introduction
Bayesian and adaptive trial designs have been used to support Food and Drug Administration (FDA) approval of drugs and medical devices and are proposed as an efficient way to achieve valid and reliable evidence from comparative effectiveness research (CER) [1 –9] as defined by the Institute of Medicine [10]. To our knowledge, there have been no Bayesian adaptive CER trials performed and just one such trial plan published [11].
We initiated a project, ‘REsearch in ADAptive methods for Pragmatic Trials’ (RE-ADAPT), funded by the National Heart, Lung and Blood Institute (NHLBI), whose aim is a proof-of-concept that Bayesian adaptive methods may have potential benefits for CER trials. RE-ADAPT will re-execute the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) [12] using patient-level data to evaluate whether a Bayesian adaptive design might have accomplished the original ALLHAT objectives more efficiently in terms of the number of patients enrolled and the calendar time of the trial.
The aim of the RE-ADAPT study is to emulate the actual process of designing a Bayesian adaptive design tailored to the original aims of the ALLHAT trial (as opposed to performing a Bayesian reanalysis of ALLHAT). The aim of this article is to describe in detail the design process of the simulation protocol. We describe a systematic review of pre-ALLHAT literature and derivation of priors to guide the designs, the specific designs that will be considered, the adaptive mechanisms (e.g., adaptive randomization) that are considered, the criteria on which adaptation decisions will be based, and factors on which the designs will be compared (e.g., final allocation of patients, duration of trial, total sample size, and final conclusions).
By prospectively publishing our protocol before we execute our new designs, we hope to address possible concern of hindsight bias – simply choosing the best design from a set of designs that would have led to a more efficient trial. To further reduce potential/concern for bias, during the design process, the two primary designers (JTC and KRB) remained blinded to the ALLHAT dataset, did not read the clinical outcome articles, and relied only on the original ALLHAT protocol and clinical discussions with cardiovascular experts regarding what cardiologists and trial designers would have known prior to the design of ALLHAT. These discussions, as well as input from the ALLHAT clinical trials center director and statistician (B.R.D.), were used to design our simulation study as though it were occurring historically at the time of ALLHAT.
ALLHAT was selected as a case study because it was large, nationally prominent, evaluated active comparators within community care settings, a public-use patient-level dataset [13] was available, was costly (US$135 million) [14], and was sufficiently lengthy (8 years) that practice patterns and thus clinical questions (e.g., combination versus monotherapy becoming more standard) may have changed during the course of the trial [15].
Co-sponsored by NHLBI and the Department of Veterans Affairs, ALLHAT enrolled 42,418 patients (aged 55 years or above, 47% women and 36% African–American) with at least one cardiovascular disease risk factor besides hypertension [16] to compare three newer antihypertensive medications to a diuretic for reducing fatal coronary heart disease (CHD) and nonfatal myocardial infarction (MI) [3,4,12]. ALLHAT incorporated several adaptive design features including conditions for early study termination and arm dropping [17]. ALLHAT monitored each comparison with its own monitoring guideline with alpha = 0.027 according to the Dunnet procedure. Early success and early futility were both based on stochastic curtailment. The number of looks depended on the information times and the calendar times of the Data and Safety Monitoring Board (DSMB) meetings. In the ‘Discussion’ section of this article, we contrast the adaptive features employed by the ALLHAT investigators with those we have designed.
The RE-ADAPT simulation protocol
The RE-ADAPT protocol consists of developing a series of potential redesigns of ALLHAT followed by simulating them to measure efficiency and performance using de-identified ALLHAT patient data. There are five steps to the process:
Conducting a systematic literature review and derivation of priors based on literature existing when ALLHAT was designed to provide the data upon which prior evidence distributions could be derived.
Identifying possible adaptations that may improve trial efficiency involves specifying possible adaptive features (e.g., adaptive randomization or arm dropping) that might be included in such a trial.
Constructing candidate set of Bayesian adaptive designs for ALLHAT by selecting combinations of priors distributions and specific adaptive features from steps 1 and 2. This includes prespecifying designs, including timing and frequency of interim analyses when adaptations may occur, and thresholds (e.g., early stopping bounds).
Selecting an optimal design for implementation from those developed in step 3. A total of 1000 replications of each design created in step 3 were simulated under different scenarios (e.g., no difference between treatments, different effect sizes). The design providing the best operating characteristics (e.g., smallest sample size, shortest trial duration, highest power, and most patients randomized to better therapy) over the widest range of possible efficacy scenarios is chosen for implementation.
Executing all designs using actual ALLHAT data to assess the performance of the chosen optimal design and all others considered, comparing each with the original ALLHAT design. Whereas, in reality, a single design must be chosen for implementation, since this is a simulation exercise, we can/will execute the chosen design as well as those we opted ‘not’ to implement.
Details of these steps are explained in the following sections. The first 4 items have been completed; we describe the fifth prospectively.
Systematic literature review and derivation of priors
Priors are required for the Bayesian analysis. They can be classified as non-informative (‘vague’) roughly corresponding to classical analysis in which only new trial data inform the inference; or ‘informative’ (‘historical’) where the evidence distribution is formally incorporated with the new trial data.
We originally planned to create one set of designs with non-informative priors and another using historical priors for each of the four drugs under study. Our formal literature review, however, revealed that no such studies using the ALLHAT primary end point were available for any of the three comparators (angiotensin-converting enzyme (ACE) inhibitors, calcium-channel blockers (CCBs), and alpha-blockers). Therefore, the historical prior effectively matched the non-informative prior for the three comparator drugs, and we chose to incorporate only designs using non-informative priors for all four drugs.
Identify possible adaptations that may improve trial efficiency
Three types of adaptations are considered: adaptive randomization, arm dropping, and early stopping. Adaptive randomization and arm dropping may occur in the accrual stage. All designs allow early stopping of the trial for futility or success, either of which may occur in the accrual or follow-up stages (criteria for early stopping are discussed in detail in the section ‘Early Stopping of the Trial for Success or Futility’). Adaptive randomization and arm dropping (during accrual) serve two key purposes: they increase the probability that patients randomized later in the trial receive a beneficial therapy; and they can increase statistical power by prioritizing data gathering for treatments where the research question remains more uncertain. Furthermore, by performing multiple prospectively defined interim analyses during accrual and follow-up phases, the trial may stop early if primary goals are met or it becomes evident that additional information is unlikely to lead to a significant conclusion.
The following sections describe the different rules with which adaptive randomization, arm dropping, and study termination may be incorporated into designs. These rules involve predetermined thresholds that govern when and which adaptations would be made. These were determined based on simulations testing a range of potential thresholds to find those values offering the most beneficial trade-offs. The statistical and clinical benefits of designs based on the various potential thresholds were discussed between the statistical designers and the clinicians involved to replicate the actual trial design process. This included discussing the overall operating characteristics and also discussing many individual trial simulations to illustrate how the trial would proceed and the nature of possible realizations and the adaptations that would result. This, like an actual adaptive trial design, was an iterative process between the statistical team and clinical team.
All thresholds/decision points were based upon simulation and chosen before the lead statisticians acquired the ALLHAT data. Thresholds for early success stopping were chosen to conserve Type I error to less than 2.5% (one-sided). Thresholds for arm dropping were chosen to balance the probability of correctly dropping a poorly performing arm with incorrectly dropping an arm that was performing poorly early due to natural variability. Thresholds were also chosen to optimize power, trial duration, and percentage of patients randomized to the best therapy. The process of choosing thresholds is analogous to choosing a cutoff threshold for a diagnostic test when weighing sensitivity and specificity – higher, more aggressive values will lead to correctly stopping a trial early or dropping a poor arm sooner, but will also lead to increased Type I errors or an increased likelihood of erroneously dropping efficacious arms.
These decisions are subjective, and different designers and clinicians may have chosen other values. This is similar to trial designers choosing more or less aggressive stopping boundaries in a group sequential trial to match the clinical situation. For instance, we simulate data from the five scenarios discussed below (not ALLHAT data but plausible scenarios) and tuned the thresholds to behave well over this range of plausible ‘truths’. Once the values/decisions points are set, the real ALLHAT data will be used to execute the trial. Another example is provided below in the section describing adaptive arm dropping.
Adaptive randomization
Trials with adaptive randomization begin with an initial assignment probability for each study arm and are later updated at predetermined times based on ‘real time’ observed treatment effects. ALLHAT used fixed randomization with a greater proportion of patients allocated to the diuretic arm to increase power for each pair-wise comparison. We will compare this original fixed randomization approach to two alternative approaches: randomize patients proportional to the probability that each comparator arm offers the higher probability of being the best arm (probability-weighting), and randomize patients proportional to both the observed treatment effects and the uncertainty in those treatment effects (information-weighting). In both cases, randomization to the diuretic arm remains fixed at one-third of patients since this is considered standard treatment to which others are compared. Due to the low incidence of the primary end point (fatal CHD + nonfatal MI), randomization probabilities will first be updated after the 10,000th patient is enrolled and then again every 3 months until the end of accrual. Starting adaptive randomization at 10,000 patients, like other thresholds, was chosen based on comparing the operating characteristics of a variety of alternatives (e.g., 20,000 patients).
The three randomization schemes explored are as follows:
Fixed randomization (reference case): Patients are randomized according to the original allocation rules in ALLHAT throughout enrollment: 36.55% to diuretic and 21.15% to each of the three comparator arms [12].
Probability-weighted adaptive randomization: Beginning with the 10,000th patient and every 3 months thereafter, randomization probabilities are updated to be proportional to the probability that each comparator offers the best (lowest) hazard ratio (HR) compared to diuretic. Probabilities are derived from posterior distributions of HRs of each treatment at interim analyses. Thus, if all comparators have similar posterior distributions, then randomization to non-diuretic arms would occur with approximate equal probability, and the more dramatic the benefit to a particular arm, the higher the randomization probability to that arm.
The result is that the comparator arms performing better will receive more patients and overall event rates will be lower than with fixed randomization [18 –22]. However, statistical power for the comparison of the best two arms in a multi-arm trial is increased since the comparator arms of most interest receive larger numbers of patients [23].
Information-weighted adaptive randomization: This approach is similar to the probability-weighted approach, but further incorporates the precision of the HRs in the derivation of revised randomization ratios. Thus, in addition to favoring arms with the lowest observed event rates, this approach also prioritizes arms where precision is lowest, and hence, the need for additional data is highest. For example, if CCBs and alpha-blockers appear to be equally efficacious but there is greater variability surrounding the estimate for alpha-blockers, more patients would be randomized to that arm in the next cohort in order to refine its estimate. Adaptive randomization here will tend to be less aggressive than with probability-weighting [24 –26].
Many have criticized adaptive randomization [27,28] for its lack of power compared to fixed randomization. However, these criticisms focus on the two-arm case and are not relevant to this four-arm trial. We acknowledge that 1:1 randomization tends to optimize power in the two-arm case. However, as Berry [23] describes, adaptive randomization tends to increase study power for trials of three or more arms. Furthermore, he suggests that we tend to do two-armed trials because it is hard and expensive to do multi-armed balanced trials, but then having limited most of our thinking to two-arm trials, we criticize adaptive trials in the two-armed setting, which is clearly not where they shine brightest.
Adaptive arm dropping
Another adaptation type that enhances treatment allocation is arm dropping (i.e., suspension of enrollment). This can be viewed as an extension of adaptive randomization in which one or more arms are assigned a zero probability of randomization. The following four adaptive arm-dropping approaches are explored:
No arm dropping (reference case) whereby enrollment is never stopped completely, but if the adaptive allocation is allowed in the design, randomization ratios can become very small, effectively zero, if one or more of the CCB, ACE inhibitor, and alpha-blocker arms is performing poorly compared to the others.
Posterior probability-based adaptive arm dropping extends designs with adaptive randomization by suspending enrollment into arms with low randomization probabilities. This threshold is set at 0.05 with probability-weighted adaptive randomization and 0.10 with information-weighted adaptive randomization. If randomization probabilities fall below these thresholds, accrual to the arm is suspended, and the remaining arms will receive a proportional increase in randomization. At the next interim analysis, the suspended arm may resume accrual if randomization probabilities increase above the thresholds. Patients in the suspended arms continue treatment and follow-up as usual.
Arm dropping can be incorporated in the design even if randomization ratios are fixed. Instead of basing the decision to discontinue enrollment on randomization probabilities, adaptation is based on the posterior probability of effectiveness – that is, the probability that the HR of a comparator arm to the diuretic is less than 1. If this probability drops below 0.2, enrollment into the arm is stopped without possibility of resuming and both treatment and follow-up also stops.
For the 20% threshold, we looked at individual simulations and considered the frequency with which beneficial arms were erroneously terminated (due to natural variability and usually occurring at early interim analyses) versus the proportion of terminations that occurred to truly inferior arms. The higher (more aggressive) this threshold, the greater the likelihood of both good and bad arms being terminated. Therefore, this value was chosen to most often suspend poorly performing arms while rarely suspending better arms that were just at a random low. No formal utility rule was used in the decision process.
Predictive probability-based arm dropping is employed when arm dropping can occur, but is based upon predictive probabilities of trial success. In this approach, no adaptive randomization is used, and control arm randomization is fixed at 1/3 with the remaining 2/3 being divided equal between the remaining available arms. This decision rule is based upon predictive power that incorporates data observed (at the time of the interim analysis) and data likely to be observed if each arm stays in the trial. An arm is terminated if predictive power versus diuretic is ever less than 10% and patients who would have been randomized to that arm are redistributed to the remaining arms [29].
Early stopping of the trial for success or futility
Early stopping of the trial for success or futility, a feature of all the designs, will be assessed at interim analyses during the accrual and follow-up phases. Up to nine early stopping interim analyses are planned: when the 20,000th, 30,000th, and 40,000th patients are enrolled (i.e., the latter being the end of accrual), and then at 9, 18, 27, 36, 45, and 54 months after the end of accrual, as noted in Table 1. Final analysis occurs 60 months after final patient enrollment. Early stopping for futility may occur at any interim look, but early stopping for success begins at the 30,000th patient look. Even with 20,000 patients accrued, few events are observed and there is large variability in the observed treatment effects. Thus, early success stopping is not allowed at the first interim analysis.
Planned interim analyses for potential early stopping
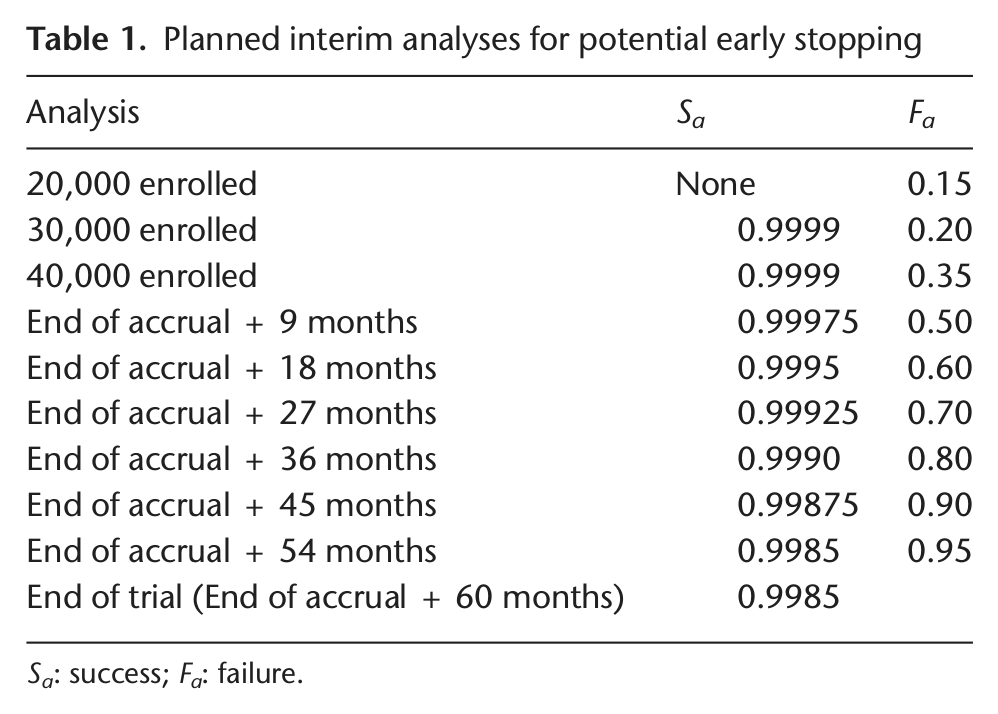
Sa : success; Fa : failure.
This serves to control Type I error rate in two ways. First, initiating adaptive randomization after 10,000 patients are enrolled but not allowing early success stopping until the 30,000-patient analysis eliminates the possibility of a Type I error at the early analyses. Meanwhile, all design variants that include adaptive randomization will increase the number of patients on arms performing best at that point in time. Thus, these arms will be more rapidly ‘tested’, and if we are truly observing a random high, more patients will be randomized to those arms and we will observe regression to the mean more rapidly, thus decreasing the likelihood of a Type I error at a subsequent analysis. If the effect is real, additional patients to the most effective arm will increase power between the best arm and the comparator.
At each early stopping analysis, the comparator arm with the best (lowest) HR is compared to the diuretic arm, and the posterior probability that the HR is below 1 is compared to the stopping boundaries for success (Sa ) and failure (Fa ) (Table 1). The trial is stopped early for success if this probability exceeds Sa , and for futility if this probability is below Fa .
Therefore, as soon as one comparator meets a stopping criterion, the trial stops. Two or all three comparators could cross a threshold simultaneously, in which case it would be reported that two or all comparators offer a significant improvement compared to diuretic. Similarly, the best comparator might cross a stopping boundary with the second best close, but not quite, achieving statistical significance. The trial would nevertheless stop as the goal is to most rapidly identify an alternative to diuretic that offers improvements on the primary cardiac outcome.
Stopping boundaries are identical across all designs. Success criteria (Sa ) have been calibrated to adjust for both multiple comparisons and frequent interim analyses such that the overall one-sided Type I error rate is less by 2.5%.
Construct candidate set of Bayesian adaptive designs for ALLHAT
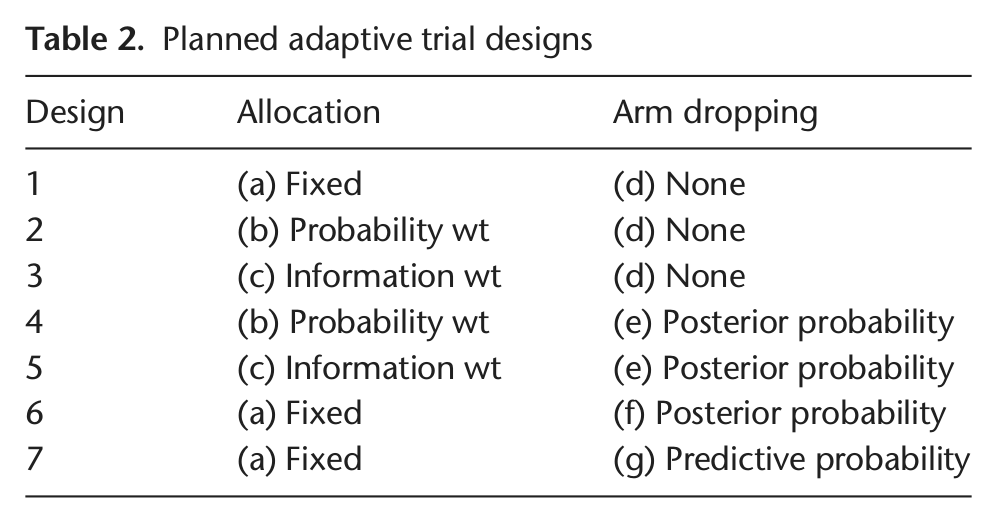
Seven different adaptive designs are created by combining the three randomization types with the three-arm-dropping approaches, as noted in Table 2. These include each of the three randomization schemes (none, probability-weighted, and information-weighted) paired with each of the three-arm-dropping schemes (none, posterior probability based, and predictive probability based). The predictive probability arm-dropping scheme incorporates the distribution of outcomes for future subjects, which is dependent upon their randomization assignments. Therefore, this strategy is more complicated if randomization assignments for future subjects vary. Consequently, the predictive-probability-based arm-dropping approach was used only in the context of fixed randomization leaving a total of seven designs.
Planned adaptive trial designs
Select optimal design for implementation
After the seven candidate designs (each individually optimized via simulation) were identified, we compared them to one another by simulating trials across a variety of plausible effectiveness scenarios. This involves testing each design via simulation to understand the potential performance measured in terms of expected (mean) sample size and duration of the trial, power, probability of stopping early for success or futility, and proportion of patients randomized to the best therapy for five different efficacy scenarios:
Null: no comparators arms better than control;
Alternative: all equally better than control (HR = 0.837);
One Works: one better than control (HR = 0.837) and the other two equal to control;
Better & Best: one best (HR = 0.837), one slightly better (0.9185), and one equal to control;
Worse: all are equally worse than control (HR = 1/0.837 =1.195).
These scenarios are based on a range of plausible effectiveness scenarios including the null and alternative hypotheses from the original ALLHAT designs and other variants on these two scenarios. In this manner, we seek to identify an optimal design: one which offers the best trade-off of highest power and most likely to terminate a futile trial early, to identify a successful treatment fastest, and to randomize the highest proportion of patients to the best treatment, all while maintaining Type I error control.
Simulating 1000 trials from each scenario for each of the seven designs produces operating characteristics for each scheme (Table 3). Note that power in the null scenario is the one-sided Type I error rate, and controlled at less than 2.5% (Table 3). In addition to studying average operating characteristics, understanding the range of possibilities for single trials is important. Numerous individual simulated trials were shown to the clinical team. Additionally, Figures 1 and 2 show distributions for study duration and the proportion of patients within each simulation randomized to the best treatment (according to the primary end point). Because each design variant uses the same stopping rules (the key differences are randomization assignment algorithms), trial durations do not change drastically across designs.
Operating characteristics for planned adaptive trial design variants
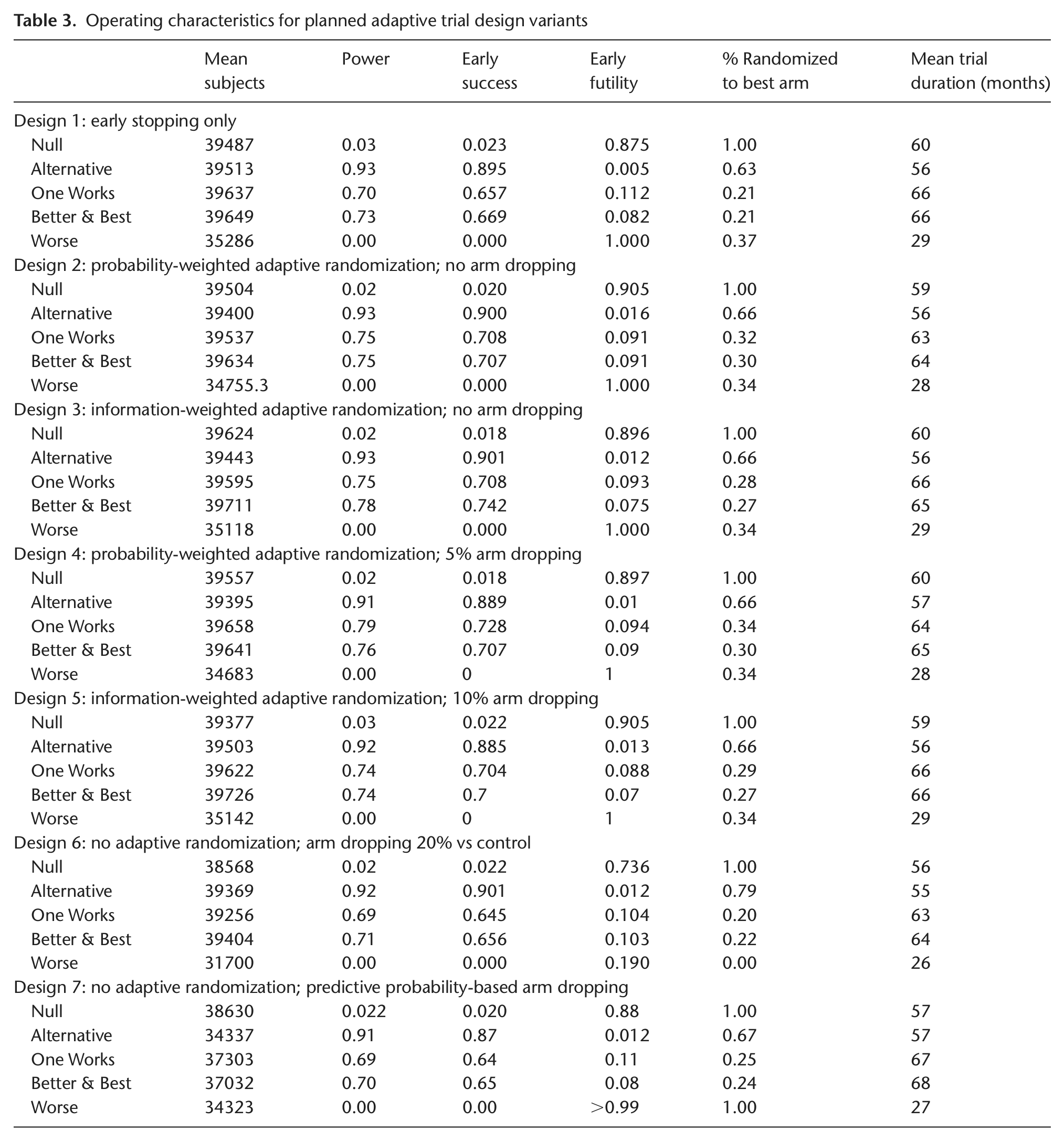

Distribution of trial duration (in years) for each design/scenario combination.
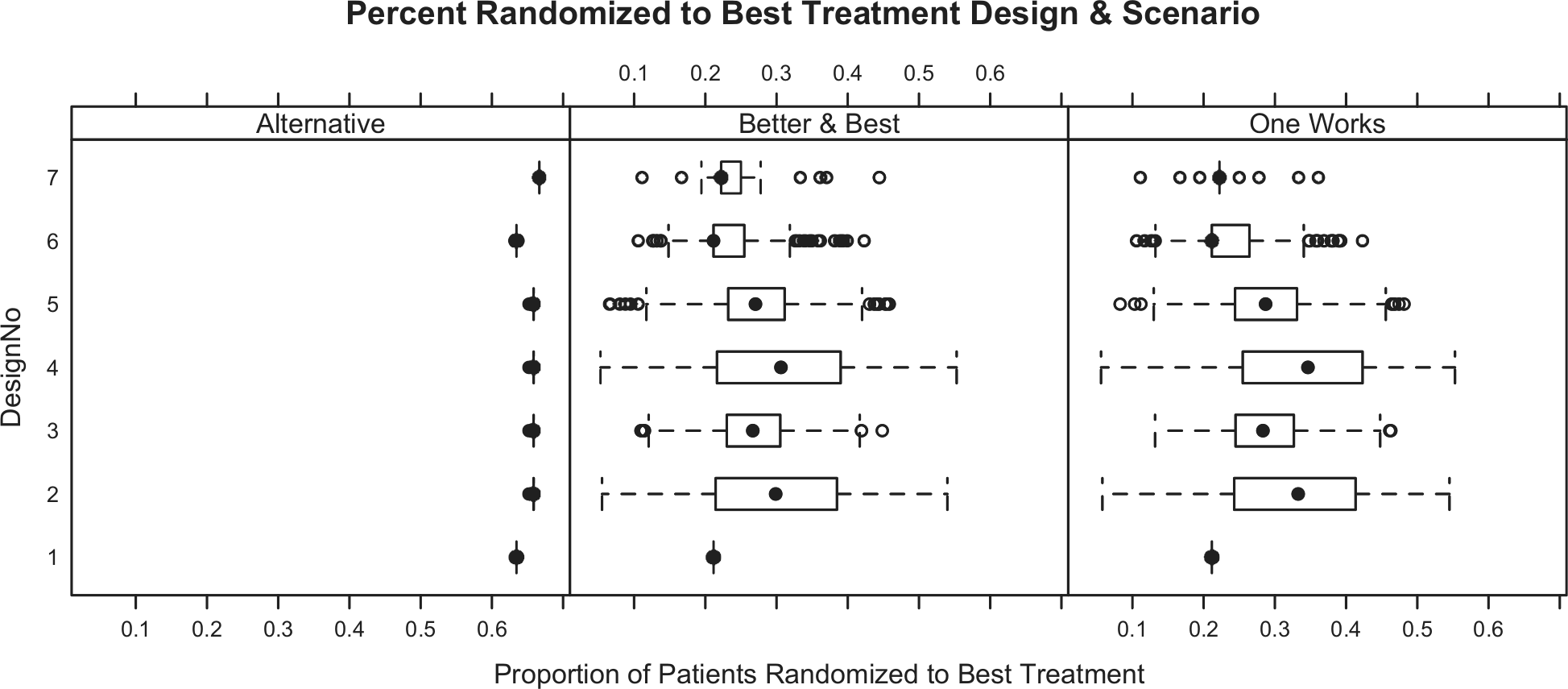
Distribution of proportion of patients randomized to the best therapy (according to primary end points) for each design/scenario combination. The null scenario (all doses equal) and scenario where all comparators are worse than control are not shown since they are the same for all designs (100% and 33%, respectively).
After considering operating characteristics of all candidate designs, we chose Design 4 (probability-weighted adaptive randomization, and probability-based adaptive arm dropping) as the design we would have implemented. Its power is high (91%, 79%, and 76%) for each scenario with an effective comparator. Design 4 offers 79% power versus 70% for a fixed randomization trial when just one comparator is superior to control and 76% versus 73% when one is clearly better and another is in between (one better, one best). In scenarios where one treatment is preferable (One Works and Better & Best), a greater proportion of patients are randomized to the better arm (34% and 30%) compared to trials with fixed randomization (including the original design) in which 21% of patients are randomized to the superior treatment. In the ‘Better & Best’ scenario, 14%, 21%, and 30% of patients are randomized to the inferior, middle, and best comparator, respectively.
The effect of adding an arm-dropping component to adaptive randomization is small but important. Without arm dropping, poor arms are eventually dropped as their probability of being the best arm approaches zero. With arm dropping, these probabilities are truncated to zero anytime they are less than 5%, and then the randomization probabilities are redistributed, thus the arm-dropping component is a bit more aggressive in assigning patients to better performing arms. For instance, when comparing Design 2 with Design 4, 2% more patients are assigned to the best therapy in the One Works scenario producing an increase in power from 75% to 79%.
This highlights the double benefit of adaptive randomization: a higher proportion of patients randomized to the more effective therapy and by the end of the trial resources (patients) are being assigned to the treatment that increases statistical power and randomized away from treatments (the one equal to control) for which we do not desire an increase in statistical power. The Technical Appendix A includes a full description of each design, including additional operating characteristics for each design. An example trial of Design 4 is also detailed there and illustrates how this design would be conducted.
Execution of Bayesian adaptive designs
The first four steps have been completed and are described in this article and in Technical Appendix A. The final step is to execute the chosen Bayesian adaptive design using the actual ALLHAT data.
We believe the best way to judge a trial (adaptive or fixed) is by simulating the trial’s conduct over a broad range of potential scenarios (efficacy scenarios, accrual rates, patient populations, etc.) and studying which trial offers the best operating characteristics over the broadest range of deviations from the primary trial assumptions. However, we understand that simulation, particularly to many clinicians, is sometimes not the most convincing tool. Therefore, until a large-scale CER trial is performed using the adaptive techniques described here, we will implement each of our seven proposed designs using the original ALLHAT data to illustrate how an actual Bayesian adaptive CER trial would likely proceed and how the inferences that result will compare to the traditional alternative (the actual ALLHAT trial).
In addition to executing Design 4, we will also execute the other six designs to understand how each would likely have performed had it been chosen. Actual ALLHAT data will be used to replicate the trial conduct including enrollment, randomization, follow-up, and end-point ascertainment. Should one comparator arm perform particularly well, the adaptive randomization process may call for more patients than were contained in its original ALLHAT arm, in which case we will resample subjects in a bootstrap fashion.
To ensure proper timing for interim analyses, special attention will be given to ensure simulated time reflects actual time between enrollment and timing of end points. Thus, only end points that had occurred and were recorded and available for analysis may inform an interim adaptation. Accumulated data at each prespecified interim looks will be analyzed to test criteria for adaptation or early trial termination. Resulting changes are incorporated and applied to the next patient cohort of enrollees. This virtual process continues until an early stopping criterion is met or the study reaches its administrative end defined by its maximum sample size and maximum follow-up time.
Evaluation criteria
Simulated outcomes of each scheme will be compared in terms of total sample size, trial duration, percentage of patients to each arm, and trial conclusions – how each trial’s inferences compare to the original ALLHAT design’s inferences. We will also look at total numbers of events, both primary and secondary, across all arms in the different designs and we will identify which components of the adaptive trial are leading to benefits or drawbacks compared with the original ALLHAT design.
Discussion
This article illustrates the redesign process we developed and will employ to simulate ALLHAT as a Bayesian adaptive trial. ALLHAT was chosen strictly for convenience as a proof of concept case study. The overall RE-ADAPT project includes several additional components presently under consideration but not addressed here, including an economic analysis of efficiency differences (e.g., trial duration or size) we may detect, and demonstrating how a combination therapy arm could have been added (if desired) during the course of the ALLHAT trial.
In this article, we describe the selected adaptive schemes we will use, stopping rules, and randomization probability updates that may increase power by allocating more patients to better performing study arms, possibly decrease trial duration and sample size, and improve other aspects of trial efficiency. We intentionally make public our protocol prior to executing the reanalysis.
Typically in designing adaptive trials, several candidate designs are developed and tested via simulation based upon a range of possible outcome parameters and one design is selected for implementation. Selecting a single adaptive design in RE-ADAPT is not necessary, however, since the actual ALLHAT data are available and, thus, all candidate designs can easily be executed, including a variant that corresponds closely to the original design of the study. However, we do identify one preferred design that our study group would have implemented if an actual trial were being executed. This eliminates a multiplicity: executing all seven designs and simply comparing the best to the original ALLHAT design. Now, we will focus on one chosen design, prospectively identified, versus the original ALLHAT design.
The original ALLHAT trial was adaptive in that it offered early stopping at loosely defined times that were to be based on Data Monitoring Committee (DMC) meeting times. The early stopping strategies here are slightly more aggressive and not based on DMC meeting times. The major difference, however, is that adaptive randomization was not permitted under the ALLHAT protocol but is a focus here. This may offer increased power to the trial and offer patients, particularly those entering the trial at later stages, the opportunity to receive a better treatment. A final key difference is that our redesign focuses on one-sided tests versus ALLHAT, which was a two-sided trial. We believe, particularly in a case where a cheaper, better understood control arm is studied versus newer, more expensive comparators, that a DMC is unlikely to allow patients to be continually exposed to a comparator arm that is performing poorly merely to show it is statistically significantly worse. In this sense, we believe a one-armed trial better reflects how a DMC would behave.
The benefit of adaptive randomization is most obvious when one arm is superior to others, in which case a larger proportion of patients get randomized to the best treatment. In our pretrial simulation exercise, on average, 34% of patients are randomized to the best treatment arm when adaptive randomization and arm dropping were allowed (Design 4) compared with only 21% with fixed design (Design 1) and original ALLHAT design. Furthermore, power also increased with adaptation (79% in Design 4 vs 70% in Design 1). This occurs because in the fixed design, patients continue to be randomized to inferior treatments in the later stages of enrollment. In contrast, in Design 4, nearly all patients are randomized to diuretics and the best comparator in the later stages of enrollment. This is clinically beneficial for patients and provides increased statistical power for the primary question of interest.
While our aim is to understand how Bayesian adaptive applications may perform in CER trials, we realize that adaptive designs are situation specific, tailored to each unique clinical situation and research question. Therefore, we realize that findings from RE-ADAPT will not generalize to all CER situations that may be encountered.
The exercise described here will require some simplifications and assumptions. In reality, this decision would be based on results from interim data analyses and other factors that may be difficult to capture quantitatively. For instance, the DMC may also consider the safety profile of treatments, important secondary outcomes, or exogenous data that become available during the study. While one key task of the DMC is ensuring proper implementation of the protocol – including resulting adaptations – it may use its prerogative to make deviations to ensure patient safety (e.g., if adaptive randomization would increase randomization probability to an arm that was seeing an increase in a serious adverse event that was not part of the adaptive algorithm). We believe, however, that a DMC’s role in adaptive trials extends to ensuring that the protocol is followed unless there is strong reason to do otherwise. This means ensuring the implementation of all adaptive components. A DMC should not view protocol-defined adaptations as guidelines they may or may not choose to implement. Overruling protocol-defined adaptations leads to poorly understood operating characteristics and unknown Type I and Type II error rates.
This article discusses the primary aims of the RE-ADAPT study. Future articles will explore broader applications of Bayesian adaptive designs, for instance, by simulating the addition of new arms into the study or modeling other adaptations that were not consistent with the original aims of ALLHAT.
Limitations
Although our simulations will rely on actual patient data from ALLHAT and will emulate the original enrollment and follow-up process, the obvious main limitation is that it is a simulation and not a ‘live’ study. However, until a large-scale CER study is conducted using Bayesian adaptive trial methodologies, we hope this exercise will serve to illustrate their potential benefits and challenges. Most notably, we will not simulate the decision process a DMC may use in interpreting findings from interim analyses and approving changes in design, including early stopping. Our simulations will algorithmically apply changes based on adaptation rules without consideration of other contextual factors.
An important challenge with our study has been to omit the benefit of hindsight and knowledge gained from the results of ALLHAT in retrospectively formulating new designs for the trial. Although we went to some lengths to maintain a ‘veil of ignorance’, that effort was undoubtedly imperfect. In some instances, this ‘veil’ may have worked against our aim of making this simulation as realistic as possible.
Our goal is to re-execute these designs using the original ALLHAT data to make the designs’ outcomes directly comparable with the original findings. In some instances, however, adaptive randomization to better performing arms may call for a larger number of patients on an arm than was observed in ALLHAT. Our plan is to resample patients in these situations, and this can lead to an underestimation of the uncertainty of the results from these arms.
Finally, the ability to incorporate prior information is a fundamental advantage of the Bayesian approach, particularly in CER where we might expect high-quality phase-3 data are available on the therapies of interest. Unfortunately, data limitations precluded our planned systematic review and meta-analysis from providing historical priors on the primary outcome for beta-blockers, ACE inhibitors, and CCBs. Therefore, the designs described here used only non-informative priors. However, the sensitivity of CER trial designs to incorporation of historical information is the subject of a future article.
Conclusion
For CER to achieve its lofty aims, investment in comparative trials is needed. Since randomized controlled trials (RCTs) are expensive and time-consuming and not always tailored to achieve CER objectives, the RE-ADAPT project was initiated to test the degree to which Bayesian adaptive trial designs may be useful in increasing CER trial efficiency and utility.
Although the FDA has issued guidance documents [5,6] and both the FDA and manufacturers have gained increasing experience and acceptance of Bayesian and adaptive designs, they have not been tested in CER settings. We hope our effort will be viewed as a valid proof-of-concept of the potential for such designs to be useful for CER and will stimulate investment in them for future CER trials.
This article describes our plans for a redesigned and re-executed ALLHAT. By publishing the details of our prespecified plans, we hope to engender reader confidence that the process can be considered a reasonable approximation of what we would have done had we designed ALLHAT itself as a Bayesian adaptive trial.
Finally, we wish to emphasize that our goal is not to criticize the ALLHAT study (it also contained aims not mentioned here, for example, a statin study component that affected the design) or to claim it should have been designed differently. Rather, we chose to re-execute the ALLHAT study using Bayesian adaptive trial methods because it was a well-designed and conducted trial that provides high-quality data in a CER setting.
Footnotes
Technical Appendix A
Acknowledgements
The authors would like to thank the National Heart and Blood Institute (NHLBI) for funding the RE-ADAPT project; the Pragmatic Approaches to Comparative Effectiveness (PACE) Initiative for its role in incubating the study design concept; RE-ADAPT investigators Jaime Caro, Stephen Gottlieb, Eberechukwu Onukwugha, Fadia Shaya, and Wallace Johnson; Kert Viele and Ashish Sanil for programming assistance; Nancy Brady for manuscript preparation; Ebenezer Oloyede for program management; and Anne Samit and Candice Morales for administrative support.
Funding
The RE-ADAPT project was supported by Award Number 1RC4HL106363-01 from the National Heart, Lung, And Blood Institute (NHLBI).
Conflict of interest
The views of Dr Bryan R Luce and Dr Rachael Fleurence expressed in this article are solely theirs and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI), its Board of Governors or Methodology Committee, or those of the employers or the NHLBI.