
Abstract
Background
VEGFA is one of the most important regulators of angiogenesis and plays a crucial role in cancer angiogenesis and progression. Recent studies have highlighted a relationship between VEGFA expression and renal cell carcinoma occurrence. However, the expression level, gene regulation network, prognostic value, and target prediction of VEGFA in renal cell carcinoma remain unclear. Therefore, system analysis of the expression, gene regulation network, prognostic value, and target prediction of VEGFA in patients with renal cell carcinoma is of great theoretical significance as there is a clinical demand for the discovery of new renal cell carcinoma treatment targets and strategies to further improve renal cell carcinoma treatment efficacy.
Methods
This study used multiple free online databases, including cBioPortal, TRRUST, GeneMANIA, GEPIA, Metascape, UALCAN, LinkedOmics, Metascape, and TIMER for the abovementioned analysis.
Results
VEGFA was upregulated in patients with kidney renal clear cell carcinoma (KIRC) and kidney chromophobe (KICH), and downregulated in patients with kidney renal papillary cell carcinoma (KIRP). Moreover, genetic alterations of VEGFA were found in patients with renal cell carcinoma as follows: 4% (KIRC), 8% (KICH), and 4% (KIRP). The promoter methylation of VEGFA was lower and higher in patients with clinical stages of KIRC and stage 1 KIRP, respectively. VEGFA expression significantly correlated with KIRC and KIRP pathological stages. Furthermore, patients with KICH and KIRP having low VEGFA expression levels had a longer survival than those having high VEGFA expression levels. VEGFA and its neighboring genes functioned in the regulation of protein methylation and glycosylation, as well as muscle fiber growth and differentiation in patients with renal cell carcinoma. Gene Ontology enrichment analysis revealed that the functions of VEGFA and its neighboring genes in patients with renal cell carcinoma are mainly related to cell adhesion molecule binding, catalytic activity, acting on RNA, ATPase activity, actin filament binding, protease binding, transcription coactivator activity, cysteine-type peptidase activity, and calmodulin binding. Transcription factor targets of VEGFA and its neighboring genes in patients with renal cell carcinoma were found: HIF1A, TFAP2A, and ESR1 in KIRC; STAT3, NFKB1, and HIPK2 in KICH; and FOXO3, TFAP2A, and ETS1 in KIRP. We further explored the VEGFA-associated kinase (ATM in KICH as well as CDK1 and AURKB in KIRP) and VEGFA-associated microRNA (miRNA) targets (MIR-21 in KICH as well as MIR-213, MIR-383, and MIR-492 in KIRP). Furthermore, the following genes had the strongest correlation with VEGFA expression in patients with renal cell carcinoma: NOTCH4, GPR4, and TRIB2 in KIRC; CKMT2, RRAGD, and PPARGC1A in KICH; and FLT1, C6orf223, and ESM1 in KIRP. VEGFA expression in patients with renal cell carcinoma was positively associated with immune cell infiltration, including CD8+T cells, CD4+T cells, macrophages, neutrophils, and dendritic cells.
Conclusions
This study revealed VEGFA expression and potential gene regulatory network in patients with renal cell carcinoma, thereby laying a foundation for further research on the role of VEGFA in renal cell carcinoma occurrence. Moreover, the study provides new renal cell carcinoma therapeutic targets and prognostic biomarkers as a reference for fundamental and clinical research.
Keywords
Introduction
Renal cell carcinoma (RCC), the most common malignant tumor in the kidney, is one of the 10 most common cancers worldwide. 1 It accounts for approximately 330,000 cases of cancer worldwide, and more than 140,000 deaths annually. 2 Among the heterogeneous subtypes of RCC, kidney renal papillary cell carcinoma (KIRP) ranks second in terms of incidence, accounting for 10%–15% of RCCs, following kidney renal clear cell carcinoma (KIRC) with an incidence of 75%–80%. 3 Kidney chromophobe (KICH) is a rare subtype of RCC, accounting for approximately 4%–5% of RCCs. 4 Advanced RCC is a fatal disease, with a 5-year survival rate of only 11.7%. 5 Surgery and targeted therapy are the primary clinical therapeutic methods for RCC. 6 Relevant clinical trials have shown that the use of immune checkpoint inhibitors in combination with antiangiogenic agents is more clinically effective in patients with RCC.7,8 However, new immunotherapy-based methods also face many challenges in clinical application. There is an urgent need to identify more promising therapeutic targets and approaches for the treatment of RCC.
Vascular endothelial growth factor A (VEGFA), a critical angiogenic factor, is an important tumor-specific factor in patients with RCC and plays a crucial role in tumor angiogenesis and progression. 9 VEGFA induces the adhesion and migration of cancer cells by binding to integrin α9β1. 10 Several VEGFA inhibitors are clinically useful.11,12 Sunitinib, a vascular tyrosine kinase inhibitor of VEGF, has been approved for the first- and second-line treatment of advanced RCC. However, sunitinib can potentially cause kidney damage and cardiovascular impairment. 11 Although VEGFA is considered to be an essential tumor-specific factor in patients with RCC, the expression level, gene regulation network, prognostic value, and regulation target of VEGFA in patients with heterogeneous subtypes of RCC (KIRC, KICH, and KIRP) remain unelucidated. Therefore, it is necessary to obtain more useful information on the relationship between VEGFA expression and RCC occurrence.
In the present study, we used multiple free online databases to identify the expression level, gene regulation network, prognostic value, and regulation target of VEGFA in patients with RCC. Moreover, this study aimed to further explore the relationship between VEGFA expression and RCC occurrence, and to provide new insights into the target treatment of patients with RCC.
Materials and methods
UALCAN analysis
UALCAN (http://ualcan.path.uab.edu/analysis.html) is an online database used as a tool for tumor subgroup gene expression and survival analyses. 13 The “Expression Analysis” module of the UALCAN database was used to analyze TCGA gene expression data, and the screening criteria were set as follows: (a) gene: VEGFA; (b) dataset: KIRC, KICH, and KIRP; (c) threshold setting conditions: P-value cutoff = 0.05. The Student's t-test was used for comparative analysis.
Gene expression profiling analysis
Gene expression profiling analysis (GEPIA) (http://gepia.cancer-pku.cn/index.html) is a free online platform that provides RNA sequencing expression data of 9736 tumors and 8587 normal samples for analyzing 14 differential mRNA expression, promoter methylation, pathological stage, and correlative prognosis in this study. The screening criteria were as follows: (a) gene: VEGFA; (b) dataset: KIRC, KICH, and KIRP; (c) threshold setting conditions: P-value cutoff = 0.05. The Student's t-test was used to analyze the expression of VEGFA and the pathological stage of RCC. The Kaplan–Meier curve was used to analyze the prognosis of patients with RCC.
cBioPortal analysis
The cBioPortal (http://cbioportal.org) is an open online database used to visualize, study, and analyze cancer genetic data. 15 In our study, the analysis of genetic alterations of VEGFA and its neighboring genes (the top 50 altered neighboring genes of VEGFA) was conducted using the cBioPortal database. The screening criteria were as follows: (a) 446, 66, and 274 samples of KIRC, KICH, and KIRP, respectively, were analyzed; (b) mRNA expression z-scores relative to all samples (log RNA Seq V2 RSEM) were obtained using a z-score threshold of ± 2.0; (c) gene: VEGFA.
STRING analysis
STRING (https://string-db.org/cgi/input.pl) is an online free database used to construct protein–protein interaction (PPI) networks between target proteins. 16 In this study, we built the PPI network interaction by screening conditions with low confidence (0.150) and defined species as Homo sapiens.
GeneMANIA analysis
GeneMANIA (http://www.genemania.org) is an analysis tool for building PPIs, generating hypotheses about gene function, analyzing gene lists, and sequencing genes for function determination. 17 In this study, we constructed interaction networks for exploring the role of VEGFA and the top 50 altered neighboring genes.
Metascape analysis
Metascape (https://metascape.org) is a simple and powerful gene function annotation and analysis tool that can help users apply the currently popular bioinformatics analysis methods for the analysis of batch genes and proteins to understand gene or protein function. 18 In our study, Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of VEGFA and its altered neighboring genes in RCC were investigated using Metascape.
TRRUST analysis
TRRUST (https://www.grnpedia.orgtrrust/) is a manually curated database of human transcriptional regulatory networks that contain 8444 and 6552 transcription-factor-target regulatory relationships of 800 human transcription factors. 19 In this study, we attempted to identify the key factor regulating the expression of VEGFA and its altered neighboring genes in patients with RCC using the TRRUST database.
LinkedOmics analysis
LinkedOmics (http://www.linkedomics.org) is a public online platform that includes multi-omics data from all 32 TCGA cancer types. 20 It provides methods for analyzing and comparing cancer multi-omics data within and across tumor types. In our study, kinase target enrichment, miRNA target enrichment, and genes differentially expressed in correlation with VEGFA were conducted using the “LinkInterpreter” module of LinkedOmics. The screening criteria were set as follows: (a) a minimum number of three genes (size); (b) cancer type: KIRC, KICH, and KIRP; (c) a simulation of 500; (d) search attribution: VEGFA and the top 50 altered neighboring genes; (e) target dataset: RNA-seq (data type).
Timer analysis
TIMER (https://cistrome.shinyapps.io/timer/) is a comprehensive resource for the systematical analysis of tumor-infiltrating immune cells, including B cells, CD4+T cells, CD8+T cells, neutrophils, macrophages, and dendritic cells. 21 In our study, the correlation between VEGFA expression level and the infiltration of immune cells was assessed by the “gene module” of TIMER.
Results
VEGFA expression in patients with RCC
We compared the expression levels of VEGFA in patients with RCC having normal human tissues, and found that the transcriptional levels of VEGFA stratified by sample type, sex, and KIRC pathological stage were significantly upregulated (P < 0.001) (Figure 1(a) to (c)). Similarly, the expression level of VEGFA in KICH tissues was increased based on sample type, sex, and KICH stage 2 (P < 0.05) (Figure 1(d) to (f)). However, the expression level of VEGFA in KIRP tissues was downregulated based on the sample type and male sex (P < 0.05) (Figure 1(g) and (h)). Furthermore, we assessed the correlation between the differential expression of VEGFA and the pathological stage in patients with RCC. Our results showed a significant correlation between the expression of VEGFA and the pathological stage in patients with KIRC (P = 0.0471) and KIRP (P = 4.36e-6) (Figure 2). Moreover, we used GEPIA to evaluate the prognostic value of VEGFA expression in patients with RCC. The overall survival was longer in patients with KIRP having low rather than high VEGFA expression levels (P = 0.00044) (Figure 3(c)). The percent survival was higher in patients with KIRP having low than high VEGFA expression levels before 100 months (P = 0.0016) (Figure 3(f)). However, a contrary result was found after 100 months in patients with KIRP (Figure 3(f)).
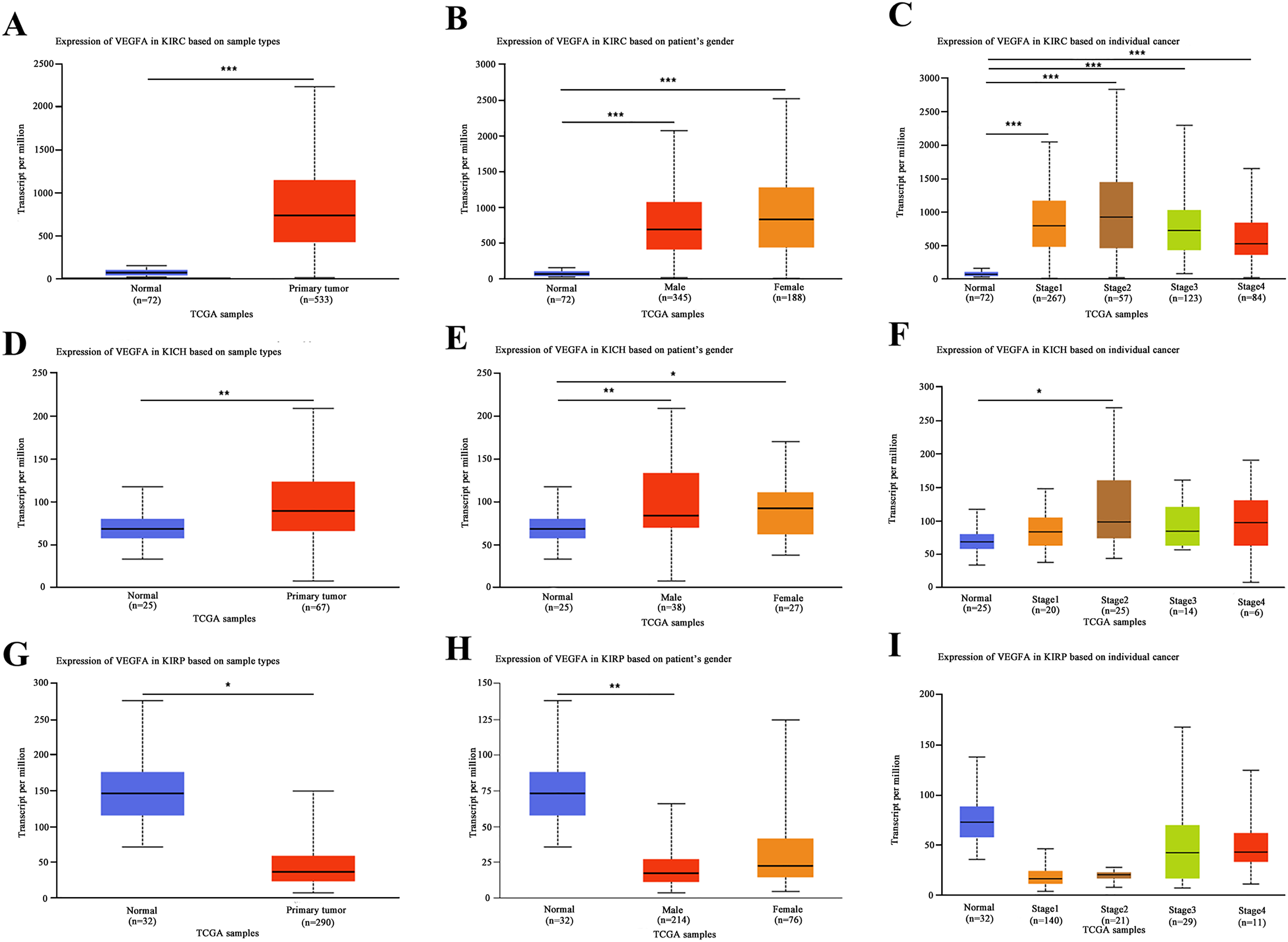
The transcription levels of VEGFA in RCC, stratified based on gender and stages (UALCAN). (a) Boxplot showing relative expression of VEGFA in normal and KIRC samples. (b) Boxplot showing relative expression of VEGFA of either gender in normal individuals and KIRC patients. (c) Boxplot showing relative expression of VEGFA of stages in normal individuals and KICH patients. (d) Boxplot showing relative expression of VEGFA in normal and KICH samples. (e) Boxplot showing relative expression of VEGFA of either gender in normal individuals and KICH patients. (f) Boxplot showing relative expression of VEGFA of stages in normal individuals and KICH patients. (g) Boxplot showing relative expression of VEGFA in normal and KIRP samples. (h) Boxplot showing relative expression of VEGFA of either gender in normal individuals and KIRP patients. (i) Boxplot showing relative expression of VEGFA of stages in normal individuals and KIRP patients. Data are mean ± SE. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
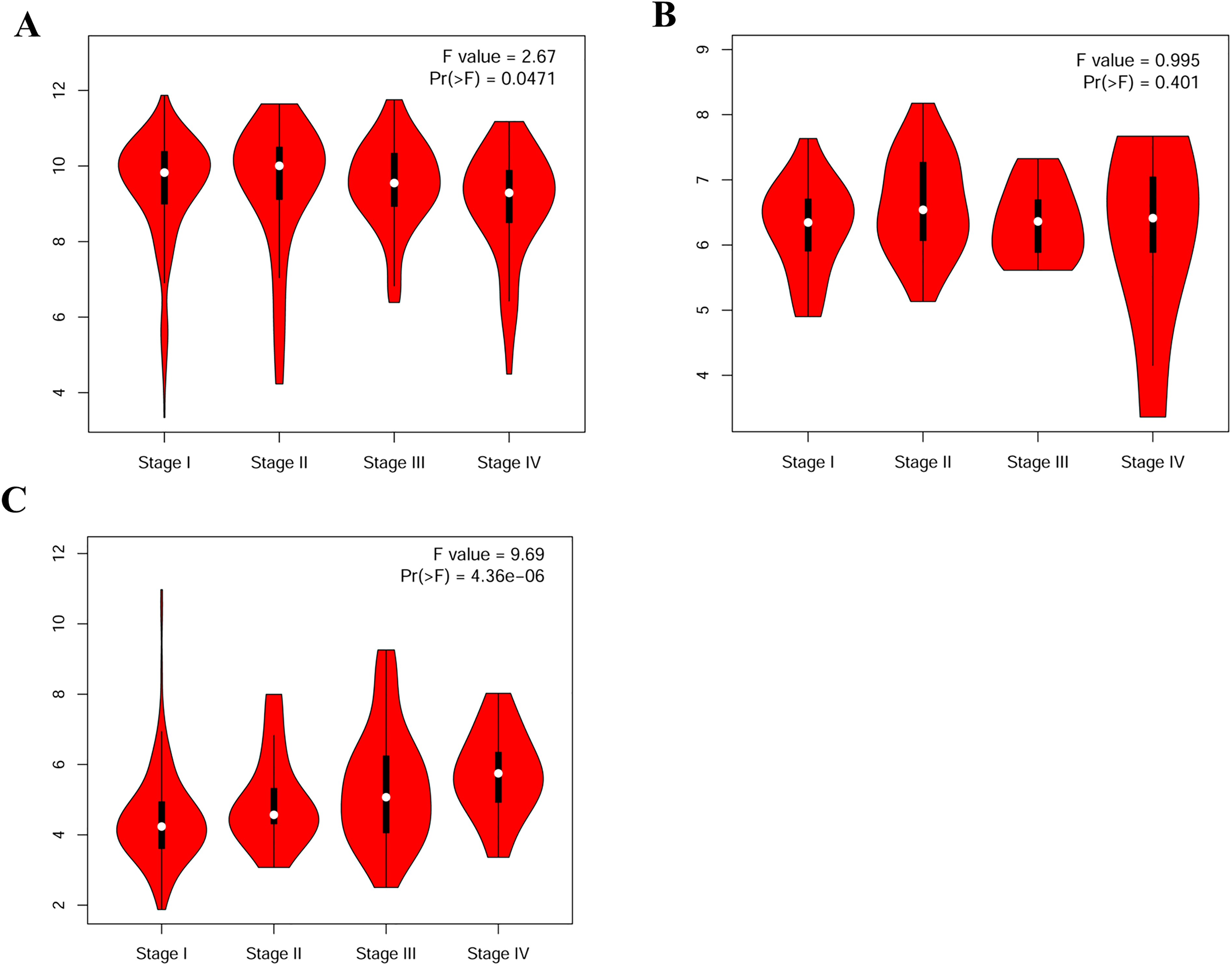
Correlation between the expression level of VEGFA and the pathological stage in RCC (GEPIA). (a) KIRC; (b) KICH; (c) KIRP.

The prognostic value of VEGFA in RCC (GEPIA). (a) The overall survival curve of VEGFA in KIRC; (b) the overall survival curve of VEGFA in KICH; (c) the overall survival curve of VEGFA in KIRP; (d) the disease-free survival cure of VEGFA in KIRC; (e) the disease-free survival cure of VEGFA in KICH; (f) the disease-free survival cure of VEGFA in KIRP.
Genetic alteration and promoter methylation level of VEGFA expression in patients with RCC
Genetic alterations of VEGFA in patients with RCC were evaluated using TCGA. Our results showed that VEGFA expression was altered by 4% in patients with KIRC (Supplementary Figure 1(a)). However, the promoter methylation level of VEGFA expression was lower in patients with KIRC than in normal humans (Supplementary Figure 1(b)). Moreover, we found an 8% VEGFA expression level alteration in patients with KICH (Supplementary Figure 1(c)). Similarly, VEGFA expression was altered by 4% in patients with KIRP (Supplementary Figure 1(e)). However, the promoter methylation level of VEGFA expression was higher in patients with pathological stage 1 KIRP than in normal individuals (Supplementary Figure 1(f)).
Neighboring gene alteration and VEGFA interaction network in patients with RCC
The neighboring gene alteration of VEGFA expression in patients with RCC was analyzed using cBioPortal software. We found VEGFA neighboring gene alteration frequencies ≥11.11%, ≥20%, and ≥9.09% (the 50 most frequently altered neighboring genes) in patients with KIRC, KICH, and KIRP, respectively (Supplementary Tables 1–3). The most frequently altered VEGFA neighboring genes in patients with KIRC were VHL (72.22%), PBRM1 (33.33%), and MUC16 (22.22%) (Supplementary Table 1). In addition, TRRAP (60.00%), PABPC1 (60.00%), and MUC5B (60.00%) were the most frequently altered VEGFA neighboring genes in patients with KICH (Supplementary Table 2). The three most frequently altered VEGFA neighboring genes in patients with KIRP were BAP1 (27.27%), MS4A15 (18.18%), and TGM7 (18.18%) (Supplementary Table 3).
Key regulated factors of VEGFA and the top 50 neighbor altered genes in RCC (TRRUST).
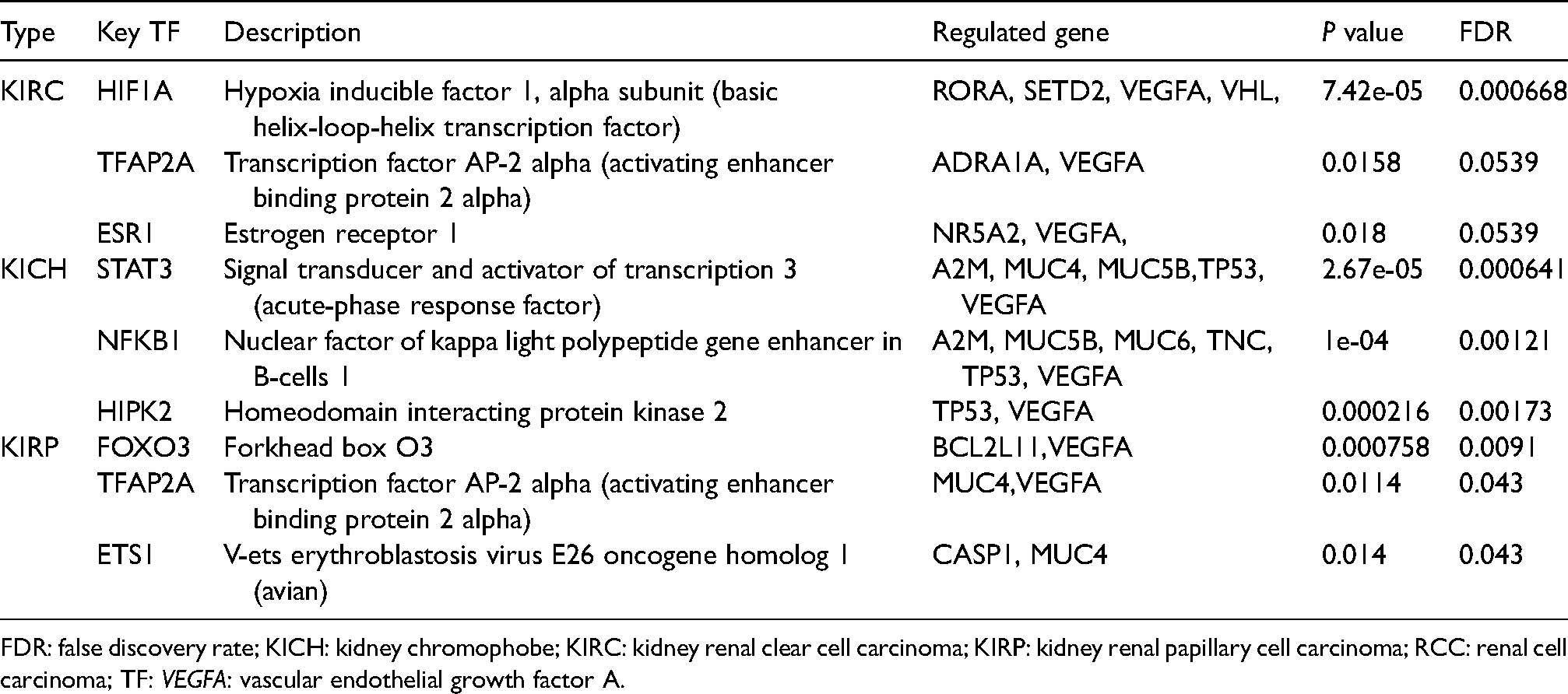
FDR: false discovery rate; KICH: kidney chromophobe; KIRC: kidney renal clear cell carcinoma; KIRP: kidney renal papillary cell carcinoma; RCC: renal cell carcinoma; TF: VEGFA: vascular endothelial growth factor A.
We then explored the potential interactions between VEGFA and its neighboring genes. The PPI network was constructed using STRING software. We obtained 43 nodes and 95 edges in the PPI networks in patients with KIRC (Supplementary Figure 2(a)). Moreover, peptidyl-lysine trimethylation, peptidyl-lysine methylation, extracellular matrix, and protein O-linked glycosylation were the primary functions of VEGFA and its neighboring genes in patients with KIRC (Supplementary Figure 2(b)). We also obtained 44 nodes and 125 edges in the PPI networks in patients with KICH (Supplementary Figure 2(c)). VEGFA and its neighboring genes had the following functions in patients with KICH: O-glycan processing, Golgi lumen, protein O-linked glycosylation, epithelial structure maintenance, and intermediate filament cytoskeleton organization (Supplementary Figure 2(d)). Furthermore, 30 nodes and 57 edges were obtained in the PPI networks in patients with KIRP (Supplementary Figure 2(e)). Muscle structure development, muscle cell differentiation, muscle tissue development, and the development of the structural constituent of muscle tissue were the primary functions of VEGFA and its neighboring genes in patients with KIRP (Supplementary Figure 2(f)). VEGFA is connected to its neighboring genes into a complex interaction network by co-expression, physical interactions, predicted shared protein domains, and co-localization (Supplementary Figure 2(b) to (f)).
GO and KEGG pathway enrichment analysis
The GO function and KEGG pathway enrichment analysis of VEGFA and the top 50 altered neighboring genes in patients with RCC were explored using Metascape software. Our results showed that the cellular components related to VEGFA and its neighboring genes in patients with KIRC were mainly involved in the basement membrane, caveola, adherens junction, sperm part, and contractile fiber part (Supplementary Figure 3(a)). Moreover, coronary vasculature morphogenesis, cardiomyocyte development, the positive regulation of axon extension, and heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules were the main biological processes of the expression of VEGFA and its altered neighboring genes in patients with KIRC (Supplementary Figure 3(b)). The molecular functions of VEGFA and its top 50 altered neighboring genes in patients with KIRC mainly included extracellular matrix structural constituent, cell adhesion molecule binding, and catalytic activity, as well as acting on RNA (Supplementary Figure 3(c)). The KEGG pathway of VEGFA and its neighboring genes in KIRC were mainly involved in the AGE-RAGE signaling pathway in diabetic complications and the PI3K-Akt signaling pathway (Supplementary Figure 3(d)). The Golgi lumen, extracellular matrix, ATPase complex, and Z disc were cellular components related to the expression of VEGFA and its neighboring genes in patients with KICH (Supplementary Figure 3(e)). In addition, the main biological processes involving the expression of VEGFA and its top 50 altered neighboring genes in patients with KICH were the maintenance of the gastrointestinal epithelium, the negative regulation of cellular component organization, and wound healing (Supplementary Figure 3(f)). The molecular functions of VEGFA and its neighboring genes in patients with KICH were ATPase activity, actin filament binding, and protease binding (Supplementary Figure 3(g)). ATP-binding cassette (ABC) transporters and human T-cell leukemia virus 1 infection were involved in the KEGG pathway analysis of VEGFA and its top 50 altered neighboring genes in patients with KICH (Supplementary Figure 3(h)). Furthermore, the cellular components related to VEGFA and its neighboring genes in patients with KIRP were mainly involved in the Golgi lumen and the centriole (Supplementary Figure 3(i)). Moreover, tissue homeostasis, myeloid cell homeostasis, O-glycan processing, and cellular response to lipids were the main biological processes involved in the expression of VEGFA and its altered neighboring genes in patients with KIRP (Supplementary Figure 3(j)). The molecular functions of VEGFA and its top 50 altered neighboring genes in patients with KIRP mainly included transcription coactivator activity, cysteine-type peptidase activity, and calmodulin binding (Supplementary Figure 3(k)). Vibrio cholerae infection, dilated cardiomyopathy, and human cytomegalovirus infection were involved in the KEGG pathway analysis of VEGFA and its altered neighboring genes in patients with KIRP (Supplementary Figure 3(l)).
Transcription factor targets of VEGFA in patients with RCC
Potential transcription factor, kinase, and miRNA targets of VEGFA in patients with RCC were acquired using TRRUST software (Table 1). Hypoxia inducible factor 1 alpha subunit (HIF1A), transcription factor AP-2 alpha (TFAP2A), and estrogen receptor 1 (ESR1) were the key transcription factors involved in the network of VEGFA and its neighboring genes in patients with KIRC (P < 0.05). RORA, SETD2, VEGFA, and VHL were regulated genes of HIF1A. In addition, TFAP2A regulated the functions of ADRA1A and VEGFA. NR5A2 and VEGFA were regulated genes of ESR1. Even more so, the key transcription factor targets involved in the network of VEGFA and its neighboring genes were signal transducer and activator of transcription 3 (STAT3), nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NFKB1), and homeodomain interacting protein kinase 2 (HIPK2) in patients with KICH (P < 0.01). The regulated genes of STAT3 were A2M, MUC4, MUC5B, TP53, and VEGFA. In addition, NFKB1 regulated the functions of A2M, MUC5B, MUC6, TNC, TP53, and VEGFA. TP53 and VEGFA were regulated genes of HIPK2. Furthermore, forkhead box O3 (FOXO3), TFAP2A (activating enhancer binding protein 2 alpha), and v-ets erythroblastosis virus E26 oncogene homolog 1 (ETS1) were the critical transcription factor targets of VEGFA and its neighboring genes in patients with KIRP (P < 0.05). Moreover, BCL2L11 and VEGFA were regulated genes of FOXO3. Additionally, TFAP2A regulated the functions of MUC4 and VEGFA. CASP1 and MUC4 were the genes regulated by ETS1.
Kinase and miRNA targets of VEGFA in patients with RCC
We also obtained the top three kinase targets and miRNA targets of the VEGFA network with LinkedOmics (Table 2). The VEGFA miRNA-target network in KIRC was associated with ATAAGCT MIR-21 (P < 0.05). Moreover, kinase targets of VEGFA were kinase ATM in patients with KICH (P < 0.05). Furthermore, kinase CDK1 and kinase AURKB were the kinase targets of VEGFA in patients with KIRP (P < 0.05). The miRNA targets of VEGFA were (TCGATGG) MIR-213, (TCTGATC) MIR-383, and (CAGGTCC) MIR-492 in patients with KIRP (P < 0.05).
The kinase target and miRNA target of VEGFA in RCC (linkedOmics).

FDR: false discovery rate; KICH: kidney chromophobe; KIRC: kidney renal clear cell carcinoma; KIRP: kidney renal papillary cell carcinoma; miRNA: microRNA; RCC: renal cell carcinoma; VEGFA: vascular endothelial growth factor A.
Correlation of differentially expressed genes and VEGFA expression in patients with RCC
We obtained mRNA sequencing data of 533 patients with KIRC in the TCGA database of LinkedOmics. As shown in Supplementary Figure 4(a), 20,158 genes were closely related to VEGFA. Among them, 11,417 and 8741 genes showed positive and negative correlations, respectively, with VEGFA expression. In addition, 50 significant genes were positively and negatively correlated with VEGFA expression in patients with KIRC (P < 0.05) (Supplementary Figure 4(b) and (c)). Moreover, the expression of VEGFA had a strong positive association with NOTCH4 (Pearson correlation coefficient = 0.7023, P = 2.076e–80) (Supplementary Figure 5(a)), GPR4 (Pearson correlation = 0.702, P = 2.63e–80) (Supplementary Figure 5(b)), and TRIB2 (Pearson correlation = 0.6972, P = 8.902e–79) (Supplementary Figure 5(c)) expressions. Furthermore, we analyzed mRNA sequencing data of 66 patients with KICH in the TCGA database, and found that 19,216 genes were closely related to VEGFA expression. Among them, 8820 and 10,396 genes showed positive and negative correlations, respectively, with VEGFA expression (Supplementary Figure 4(d)). We also identified 50 significant genes that were positively and negatively correlated with VEGFA expression in patients with KICH (Supplementary Figure 4(e) and (f)). In addition, VEGFA expression was positively associated with CKMT2 (Pearson correlation = 0.6474, P = 4.236e–9) (Supplementary Figure 5(d)), RRAGD (Pearson correlation = 0.6279, P = 1.673e–8) (Supplementary Figure 5(e)), and PPARGC1A (Pearson correlation = 0.6086, P = 5.93e–8) (Supplementary Figure 5(f)) expressions. Finally, we analyzed mRNA sequencing data of 290 patients with KIRP in the TCGA database. Furthermore, 20,023 genes were closely related to VEGFA expression. Among them, 11,130 and 8893 genes showed positive and negative correlations, respectively, with VEGFA expression VEGFA (Supplementary Figure 4(g)). We identified 50 significant genes that were positively and negatively correlated with VEGFA expression in patients with KIRP (Supplementary Figure 4(h) and (i)). The expression of VEGFA was positively associated with FLT1 (Pearson correlation = 0.8184, P = 2.909e–71) (Supplementary Figure 5(g)), C6orf223 (Pearson correlation = 0.7779, P = 4.606e–60) (Supplementary Figure 5(h)), and ESM1 (Pearson correlation = 0.7602, P = 6.983e–56) (Supplementary Figure 5(i)) expressions.
Immune cell infiltration and VEGFA expression in patients with RCC
To further reveal the relationship between immune cell infiltration and VEGFA expression in patients with RCC, we examined the correlation between immune cell infiltration and VEGFA expression using TIMER software. The expression level of VEGFA in patients with KIRC was positively associated with the infiltrations of CD8+T cells (Cor = 0.197, P = 3.36e-5), CD4+T cells (Cor = 0.301, P = 4.41e-11), and neutrophils (Cor = 0.223, P = 1.44e-6) (Supplementary Figure 6(a)). Furthermore, we analyzed the correlation between immune cell infiltration and VEGFA expression in patients with KICH. The infiltrations of B (Cor = 0.349, P = 4.39e-3), CD8+T (Cor = 0.398, P = 1.03e-5), and dendritic cells (Cor = 0.253, P = 4.18e-2) were positively associated with the expression of VEGFA (Supplementary Figure 6(b)). Finally, the expression level of VEGFA in patients with KIRC was positively associated with the infiltrations of B (Cor = 0.16, P = 1.04e-2), CD8+T (Cor = 0.179, P = 0.384e-3), and CD4+T cells (Cor = 0.157, P = 1.14e-2) (Supplementary Figure 6(c)). However, macrophage infiltration (Cor = −0.226, P = 3.22e-4) was negatively associated with VEGFA expression (Supplementary Figure 6(c)).
Discussion
VEGFA, a member of the VEGF family, is an important cytokine and a leading inducer of angiogenesis. 22 Preclinical studies have reported that VEGFA is overexpressed in patients with RCC.23,24 However, the expression level of VEGFA in patients with RCC remains controversial.25,26 Tumor angiogenesis plays a vital role in the progression of RCC. Drugs targeting tumor angiogenesis have been used in the treatment of RCC for nearly 20 years. 27 However, the therapeutic effects of these drugs are still not ideal, and their side effects need to be reduced. We sought to reveal the gene regulation network, prognostic value, and target prediction of VEGFA in patients with RCC.
Moreover, we explored the expression level of VEGFA and the correlation between the differential expression of VEGFA and pathological stage in patients with RCC. VEGFA expression was upregulated in patients with KIRC and KICH rather than in normal individuals. Nevertheless, patients with KIRP had a downregulated VEGFA expression. These findings were similar to previous study findings in patients with KIRC, KICH, and KIRP,26,28 and were contrary to previous study findings in patients with KICH. 25 We attempted to explain the conflicting results through gene alteration and promoter methylation in patients with RCC. We found that genetic alterations occurred in 4%, 8%, and 4% of patients with KIRC, KICH, and KIRP, respectively. Moreover, the promoter methylation level of VEGFA was lower in patients with different KIRC stages than in healthy individuals; in contrast, the promoter methylation level of VEGFA was higher in patients with stage 1 KIRP. However, no differences in the promoter methylation level of VEGFA were found between patients with KICH and healthy individuals. Therefore, we assume that the genetic alteration and methylation of VEGFA may be the leading cause of different VEGFA expressions in patients with RCC.
In addition, we found a significant correlation between VEGFA expression and KIRC and KIRP pathological stages. Furthermore, the survival of patients with KIRP having low VEGFA expression levels was longer than that of patients with KIRP having high VEGFA expression levels. Notably, we obtained opposite results after 100 months in patients with KIRP based on disease-free survival curves. Thus, the VEGFA expression level may be a prognostic indicator in patients with KIRP. Some studies have indicated that VEGF expression may be an essential feature of advanced RCC. The detection of CD147/VEGF co-expression may help predict the prognosis of patients with advanced RCC. 29
Moreover, the 50 most frequent VEGFA neighboring genes were altered in patients with RCC according to the following frequencies: ≥11.11% (KIRC), ≥20% (KICH), and ≥9.09% (KIRP). We then explored the potential interactions and functions of VEGFA and its neighboring genes; these were found to have complex and tight connection networks. In patients with KIRC, these genes were mainly involved in protein methylation. However, in patients with KICH, VEGFA neighboring genes functioned in protein glycosylation and cell structure maintenance. Furthermore, VEGFA neighboring genes were mainly related to muscle fiber growth and differentiation in patients with KIRP. Dynamic changes in protein methylation are essential for cell fate determination and cell development. Protein methylation occurs primarily on the side chains of Lys and Arg residues. 30 For example, histone methylation is commonly observed in cancers with methylation at Lys and Arg residues. 31 Aberrant methylation and O-glycosylation are common epigenetic tumor modifications. Aberrant O-glycosylation contributes to the development of cancer through a direct induction of oncogenic properties in cancer cells. 32 KIRP is a malignant tumor of the renal parenchyma having a papillary or tubular papillary structure with a fibrous vessel axis. KIRP occurrence may be associated with the occurrence of RCCs with angioleiomyoma-like stroma (also known as smooth muscle or leiomyoma stroma), owing to the substantial similarity of their immunohistochemical phenotypes. 33 The above evidence reveals that VEGFA neighboring genes might impact the occurrence and progression of RCC.
Furthermore, GO enrichment analysis revealed that the functions of VEGFA and its neighboring genes in patients with KIRC are mainly related to cell adhesion molecule binding and catalytic activity, as well as acting on RNA. The epithelial cell adhesion molecule (EPCAM) has recently gained attention as a candidate protein for the diagnosis, prognosis, and treatment of various tumors. EPCAM is an independent prognostic molecular marker in RCC, and may provide auxiliary information for better prognosis. 34 We found that ATPase activity, actin filament binding, and protease binding were the molecular functions of VEGFA and its neighboring genes in patients with KICH. Mitochondrial morphological and functional changes are known to occur in patients with KICH 35 ; ATPase activity alteration may be a potential pathogenic mechanism. However, further studies are required to confirm this mechanism. In our study, the molecular functions of VEGFA and its altered neighboring genes in patients with KIRP mainly included transcription coactivator activity, cysteine-type peptidase activity, and calmodulin binding. Calmodulin is an ubiquitous intracellular calcium-binding protein reportedly associated with the cell cycle. Patients with RCC are known to exhibit a positive correlation between tumor calmodulin content and tumor growth rate. 36 We found that the AGE-RAGE signaling pathway in diabetic complications, PI3K-Akt signaling pathway, ABC transporter action, and human T-cell leukemia virus 1 infection were mainly involved in the KEGG pathway analysis of VEGFA and its neighboring genes in patients with RCC; these processes are closely related to cancer occurrence and progression. 37 Thus, regulating these signaling pathways may be a potential RCC treatment target.
We also analyzed the targets and regulators of VEGFA in patients with RCC. We first explored the transcription factor targets of VEGFA and its neighboring genes in patients with RCC, and found that HIF1A, TFAP2A, and ESR1 were crucial regulatory factors in patients with KIRC. HIF-1 is a key gene regulator involved in the cellular response to hypoxia. HIF-1 overexpression is associated with RCC pathogenesis; moreover, the functional polymorphism of HIF1A may lead to susceptibility to RCC, and HIF1A polymorphism may affect RCC recurrence, progression, and survival. 38 However, TFAP2A and ESR1 have not been reported in patients with RCC. We also found that STAT3, NFKB1, and HIPK2 were significantly regulated factors in patients with KICH. STAT3 proteins are key transcription factors that are abnormally activated in a variety of malignancies, including RCC. 39 STAT3 inhibition can hinder RCC cell migration and invasion. 40 NFKB1 is related to the pathogenesis of many malignant tumors, including RCC. Functional NFKB1 promoter polymorphism is associated with an increased risk of RCC. 41 HIPK2 plays a unique role in the regulation of cell apoptosis and proliferation, as well as DNA damage repair and other basic processes; thus, HIPK2 has attracted increasing attention. FOXM1 phosphorylation by HIPK2 promotes transcriptional activity and FOXM1 cell proliferation in patients with RCC, which may be a potential RCC treatment mechanism. 42 In our study, FOXO3, TFAP2A, and ETS1 were found to be critical transcription factor targets of VEGFA and its neighboring genes in patients with KIRP. FOXO3-mediated phosphofructokinase-M expression inhibits RCC cell growth, migration, and invasion, 43 FOXO3 can be used as a new biomarker as a new therapeutic target in RCC treatment. 44 ETS1 appears to be a key transcription factor in extracellular matrix remodeling during angiogenesis; it may be involved in RCC angiogenesis. 45 We further explored the VEGFA-associated kinase targets (ATM in KICH, as well as CDK1 and AURKB in KIRP) and VEGFA-associated miRNA targets (MIR-21 in KIRC; MIR-213, MIR-383, and MIR-492 in KIRP). Over the past decade, many studies have shown that protein kinase disorders or mutations play a causal role in cancer occurrence. Cancer research has demonstrated the critical role of many protein kinases in human tumorigenesis and cancer progression, making these molecules effective candidates for new targeted therapies. 46 Kinase inhibitors have revolutionized the treatment of certain types of malignant tumors. 47 miRNAs are a class of non-coding RNAs that play an essential role in the regulation of gene expression. miRNA inhibitors reduce the expression of target genes by interacting with the 3′ untranslated region of target genes. Their important role in regulating the expression of tumor suppressor genes and oncogene expression enhances their role in tumorigenesis. 48 The discovery of miRNA targets may eventually clarify the mechanisms underlying tumorigenesis and anticancer drug development. 49 This study provides a reference and highlights the importance of further analyses of regulated factors, kinases, and miRNAs as potential therapeutic targets for RCC treatment.
We explored the correlation between differentially expressed genes and VEGFA expression in patients with RCC. More than 10,000 genes were positively and negatively correlated with VEGFA expression in patients with RCC. Among them, the genes with the highest correlation with VEGFA were found in patients with RCC: NOTCH4, GPR4, and TRIB2 in KIRC; CKMT2, RRAGD, and PPARGC1A in KICH; and FLT1, C6orf223, and ESM1 in KIRP. However, targeting these cancer-related genes may provide an adjuvant therapy for RCC. Immunoinfiltration of RCC is closely related to clinical prognosis. 50 The transportation of immune cells to cancer sites occurred mainly via blood vessels. As expected, we found that VEGFA expression in patients with RCC was positively associated with immune cell infiltration, including CD8+T cells, CD4+T cells, macrophages, neutrophils, and dendritic cells. We hope to improve the immune cell infiltration of RCCs by developing drugs acting on VEGFA or VEGFA-related regulatory targets.
In conclusion, by exploring the expression level and gene regulation network of VEGFA in patients with RCC, we have provided more insights into the study and treatment of RCC. Furthermore, we identified new therapeutic targets and prognostic biomarkers for the accurate prediction of RCC patient survival.
Supplemental Material
sj-tif-1-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-1-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-tif-2-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-2-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-tif-3-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-3-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-tif-4-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-4-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-tif-5-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-5-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-tif-6-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-tif-6-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-docx-7-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-docx-7-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-docx-8-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-docx-8-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Supplemental Material
sj-docx-9-jbm-10.1177_17246008211063501 - Supplemental material for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets
Supplemental material, sj-docx-9-jbm-10.1177_17246008211063501 for System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets by Yongli Situ, Qinying Xu, Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei and Zheng Shao in The International Journal of Biological Markers
Footnotes
Acknowledgements
This work was supported by the project of financial fund science and technology special competitive allocation of Zhanjiang (Zhanke[2010]174).
Author contributions
Zheng Shao and Qinying Xu performed data analysis work and aided in writing the manuscript. Yongli Situ designed the study and assisted in writing the manuscript. Li Deng, Yan Zhu, Ruxiu Gao, Lei Lei edited the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the project of financial fund science and technology special competitive allocation of Zhanjiang (grant number Zhanke[2010]174).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.