
Abstract
Background:
The goal of this study was to screen point mutations and deletions in APC and MUTYH genes in patients suspected of familial adenomatous polyposis (FAP) in a Brazilian cohort.
Methods:
We used high-resolution melting, Sanger direct sequencing and multiplex ligation-dependent probe association (MLPA) assays to identify point mutations, and large genomic variations within the coding regions of APC and MUTYH genes.
Results:
We identified 19 causative mutations in 40 Brazilian patients from 20 different families. Four novel mutations were identified in the APC gene and two in the MUTYH gene. We also found a high intra- and inter-familial diversity regarding extracolonic manifestations, and gastric polyps were the most common manifestation found in our cohort.
Conclusion:
We believe that the FAP mutational spectrum can be population-specific and screening FAP patients in different populations can improve pre-clinical diagnosis and improve clinical conduct.
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominant disease characterized by the presence of hundreds to thousands of adenomatous polyps throughout the colon and rectum, and it has a predisposition to colorectal cancer (FAP1, OMIM#175100). 1 Of the untreated individuals with FAP, 7% develop cancer by the age of 21 years, 87% at 45, and 93% at 50. 2 FAP is also characterized by many extracolonic manifestations, of which the most common is the congenital hypertrophy of the retinal pigment epithelium (CHRPE). Gastric polyps, desmoid tumors, osteomas, fibromas, and lipomas are common benign manifestations of the disease. 3
FAP is caused by germline mutations in the adenomatous polyposis coli (APC) gene located on chromosome 5q22.1. APC is responsible for producing a multi-functional protein that works as a tumor suppressor gene that negatively regulates the oncoprotein B-catenin. Non-functional APC leads to B-catenin accumulation in the nucleus, which upregulates the transcription of genes involved in its cell cycle progression.4,5 Germline APC mutations were identified in 30% to 85% of FAP families 6 in early studies with less sensitive methods, and about 80% to 95% of FAP families using next-generation sequencing (NGS) platform. A significant number of patients diagnosed with a less aggressive form of FAP were negative for APC mutations and carried mutations in the MUTYH gene. Thus, MUTYH mutations have been shown to induce an autosomal-recessive inherited adenomatous polyposis, also known as MUTYH-associated polyposis (MAP) (FAP2, OMIM#608456), which is clinically characterized by the presence of adenomatous polyposis of the colorectum and an increased risk of colorectal cancer. 7 The colonic phenotype of MAP mimics attenuated FAP, including a susceptibility for proximal colonic neoplasms. Colonic polyposis typically occurs when patients reach 40 years old; however, polyps and cancer can occur at earlier ages. Biallelic MUTYH mutations also have been found in individuals with the early onset of colorectal cancer (CRC).8,9
FAP diagnosis is based on clinical findings, family history, and genetic testing. Although a strong family history of FAP is shared among the patients, one-third of the cases arise from de novo germline mutations. 8 Therefore, genetic screening to identify the causal mutation is crucial, since it allows pre-symptomatic clinical surveillance and accurate genetic counseling. The goal of this study is to investigate the genetic basis of FAP in a Brazilian cohort of individuals diagnosed with FAP and its implications for clinical management.
Methods
Patients
This study evaluated individuals clinically suspected of having FAP that were admitted at the Cancer Counseling Outpatient Clinic at the Clinical Hospital of Ribeirão Preto Medical School – USP, from January 2012 until December 2017. Clinical suspicions of FAP were based on the National Comprehensive Cancer Network (NCCN) guidelines: 9 at least 10–20 cumulative colorectal adenomatous polyps, desmoid tumor, hepatoblastoma, multifocal/bilateral CHRPE, and a cribriform-morular variant of papillary thyroid cancer. As previously established, patients with >100 polyps were diagnosed with classical FAP and patients with <100 polyps were diagnosed with attenuated FAP. After a positive diagnose of the new index case, the relatives were called for genetic counseling and molecular testing. A total of 70 patients were genetically tested.
Inclusion criteria considered the clinical and molecular diagnosis for FAP and the need for signed consent to participate in the study. Family history and additional clinical information were obtained from medical records. All individual participants included in the study signed the informed consent form. This study was approved by the Ethics Committee of Ribeirao Preto Medical School, University of São Paulo (Reference Number 3969/2012).
Mutation screening using high resolution melting assay
Genomic DNA was extracted from peripheral blood lymphocytes with the Wizard Genomic DNA Purification kit (Promega).
The entire APC gene-coding region was amplified with primers that flanked each intron/exon according to Miyoshi et al. 10 Likewise, we sequenced the entire MUTYH gene-coding region using primers previously described by Lopez-Villar et al. 11 We performed a high-resolution melting (HRM) analysis to screen mutations in the APC and MUTYH genes. This methodology can identify genetic variations in the DNA sequence since it discriminates DNA sequences based on their composition, length, GC content and complementarity between the two DNA strands. 12 The HRM analysis was performed in the 7500 Fast Real-Time PCR System (Applied Biosystems) using MeltDoctor HRM Master Mix (Applied Biosystems).
DNA sequencing
DNA fragments amplified by polymerase chain reaction (PCR)-HRM were subjected to direct sequencing in an automatic capillary sequencing system ABI 3500 XL (Applied Biosystems), using the BigDye Terminator Kit, and the sequencing results were analyzed through FinchTV version 1.4.0 (Geospiza Inc. 2004–2006). The sequences obtained were compared to the reference from the GenBank NM_000038.4 and GenBank NM_001128425.1, respectively, to the APC and the MUTYH genes. For pathogenicity prediction, we used the online tools Sift, 13 PolyPhen, 14 Mutation Taster, 15 UMD-Predictor, 16 and combined annotation dependent depletion (CADD). 17 We classified as pathogenic only variants that were predicted as damaging in all of the tools.
Multiplex ligation-dependent probe amplification assay
Patients that were negative for APC and MUTYH deleterious point mutations were submitted to multiplex ligation-dependent probe amplification (MLPA) for the screening of deletions or duplications in APC and MUTYH genes. We employed the SALSA MLPA P043-C1 kit (MRC-Holland), according to manufacturer’s instructions. The results were analyzed using the software Coffalyser for MLPA data interpretation. Also, we employed a PCR-based methodology to screen for deletions in exons 4 to 16 in the MUTYH gene. 18
Statistical analysis
Statistical analysis was performed using the R commander package in R and GraphPad Prism 6. The level of statistical significance was established at P<0.05. The statistical tests applied are described in their respective results.
Results
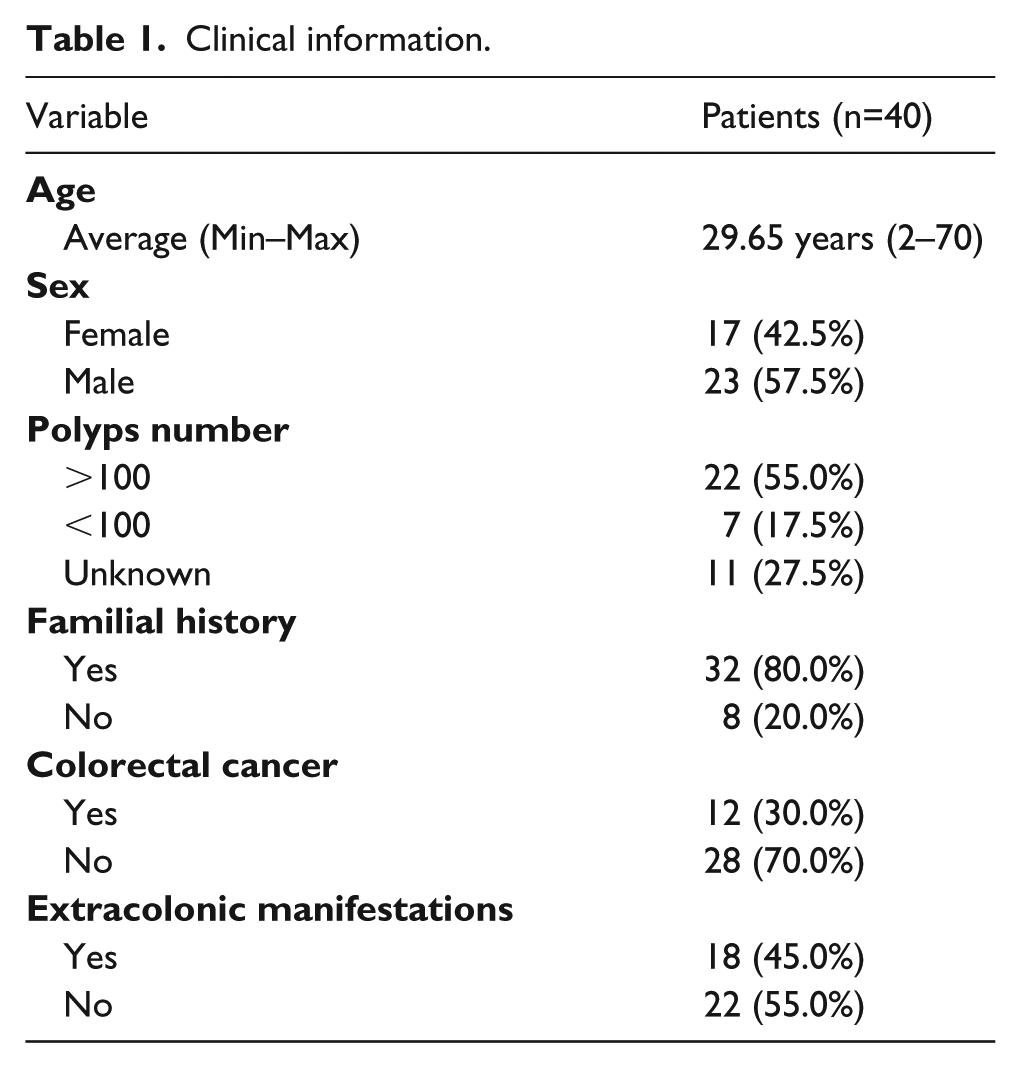
From 70 patients (belonging to 34 families) that were initially admitted to the Cancer Counseling Clinic due to the clinical criteria, a total of 40 patients (belonging to 20 families) presented molecular diagnosis for FAP and were included in the study. The majority of the families (index cases) harbored APC alterations (70%, 14/20), while the remaining 30% (6/20) carried MUTYH alterations. Among the patients, 42.5% (17/40) were female, 57.5% of were male (23/40), and the average age of onset was 29.6 years, ranging from age 2 to 70 years. In just over half of the patients (55%, 22/40), the colonoscopy detected >100 adenomatous polyps along the colon and the rectum, whereas in 17.5% (7/40) of the patients, less than 100 polyps were detected. Most of the patients (80%, 32/40) had a familial history of cancer, but only 30% (12/40) of them developed colorectal adenocarcinoma. Additionally, 45% (18/40) of the patients presented extracolonic manifestations (Supplementary Table 1). All the clinical information can be found in Table 1.
Clinical information.
APC mutations
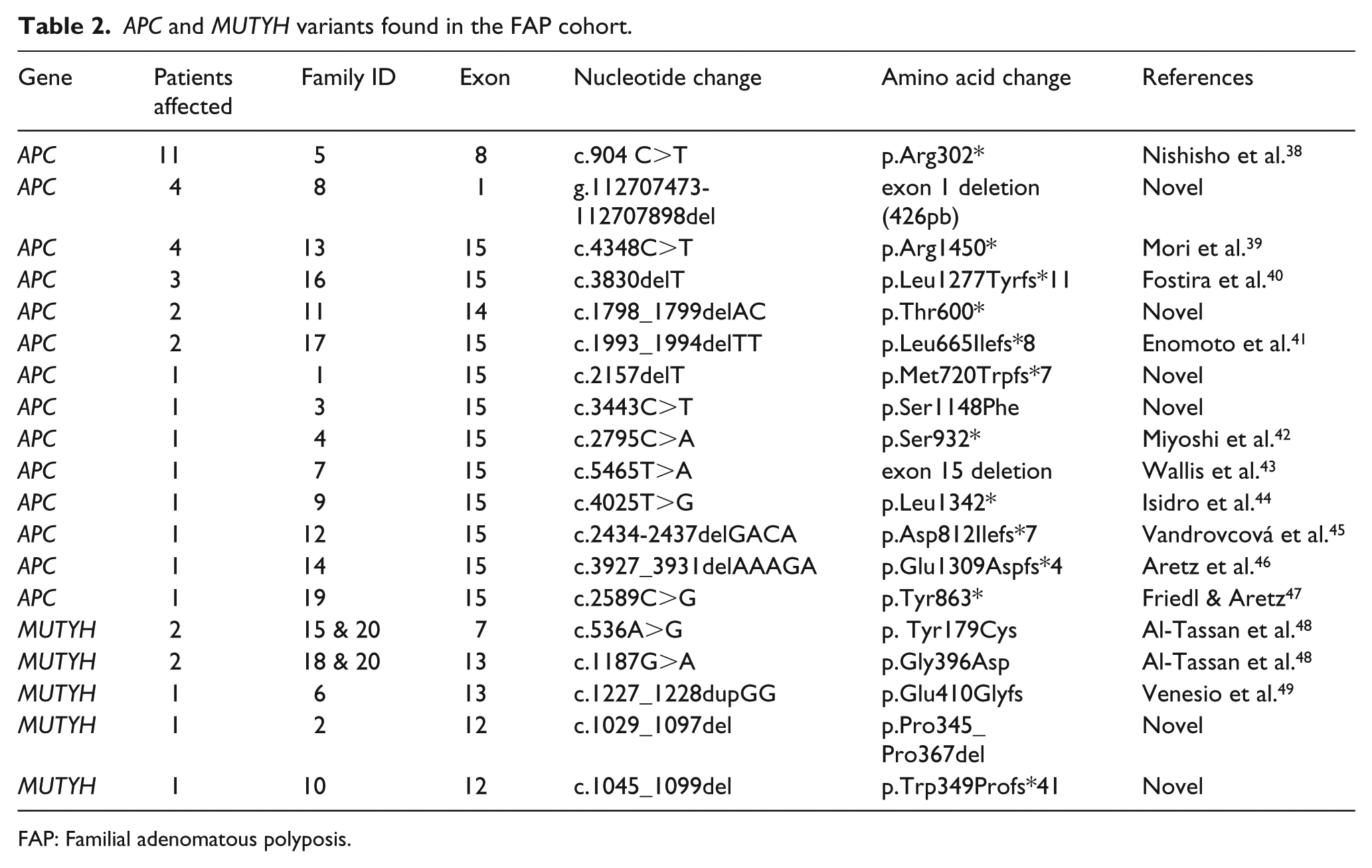
Genetic screening of APC gene identified 15 mutations in the patients: two exons deletions (exon 1 and 15), five nonsense mutations, two missense mutations, and six frameshift mutations (Table 2). As expected, members of the same family shared most of the variants (Supplementary Table 1).
APC and MUTYH variants found in the FAP cohort.
FAP: Familial adenomatous polyposis.
The majority of the mutations have been previously described (Table 2), except for four novel mutations (Figure 1(a)). One of the alterations first described here was the 426bp-deletion (g.112707473-112707898del) found in one family, which partially encompassed the promoter region and the first exon (Figure 1(a)). None of the patients carrying this deletion had any extracolonic manifestation (Family Id 8-Supplementary Table 1). Additionally, there was one frameshift, one nonsense, and one missense mutation: p.Met720Trpfs*7, p.Thr600*, and p.Ser1148Phe (Figure 1 (b–d)). The p.Met720Trpfs*7 (c.2157delT) mutation was found in one family (ID1413-Supplementary Table 1); and resulted from a nucleotide deletion at codon 720 that led to a frameshift followed by a stop codon, potentially impairing the protein function (Figure 1(b)). This patient was diagnosed at age 17 with classical FAP (>100 polyps) and had gastric polyps. The mutation p.Thr600* (c.1798_1799delAC) (Figure 1(c)) was found in two patients from the same family, both with classical FAP with early onset, diagnosed at ages 28 (ID3522-Supplementary Table 1) and 12 (ID4022-Supplementary Table 1). The third novel variant was the missense point mutation p.Ser1148Phe (c.3443C>T) (Figure 1(d)). In silico tools predicted this variant as pathogenic, with the following scores: 0.99 (PolyPhen), 0.0 (SIFT), 0.72 (Mutation Taster), 96 (UMD-Predictor), and 26.6 (CADD). This variant was found in one patient who was diagnosed at age 17 with classical FAP and who developed gastric polyps (ID1413-Supplementary Table 1).
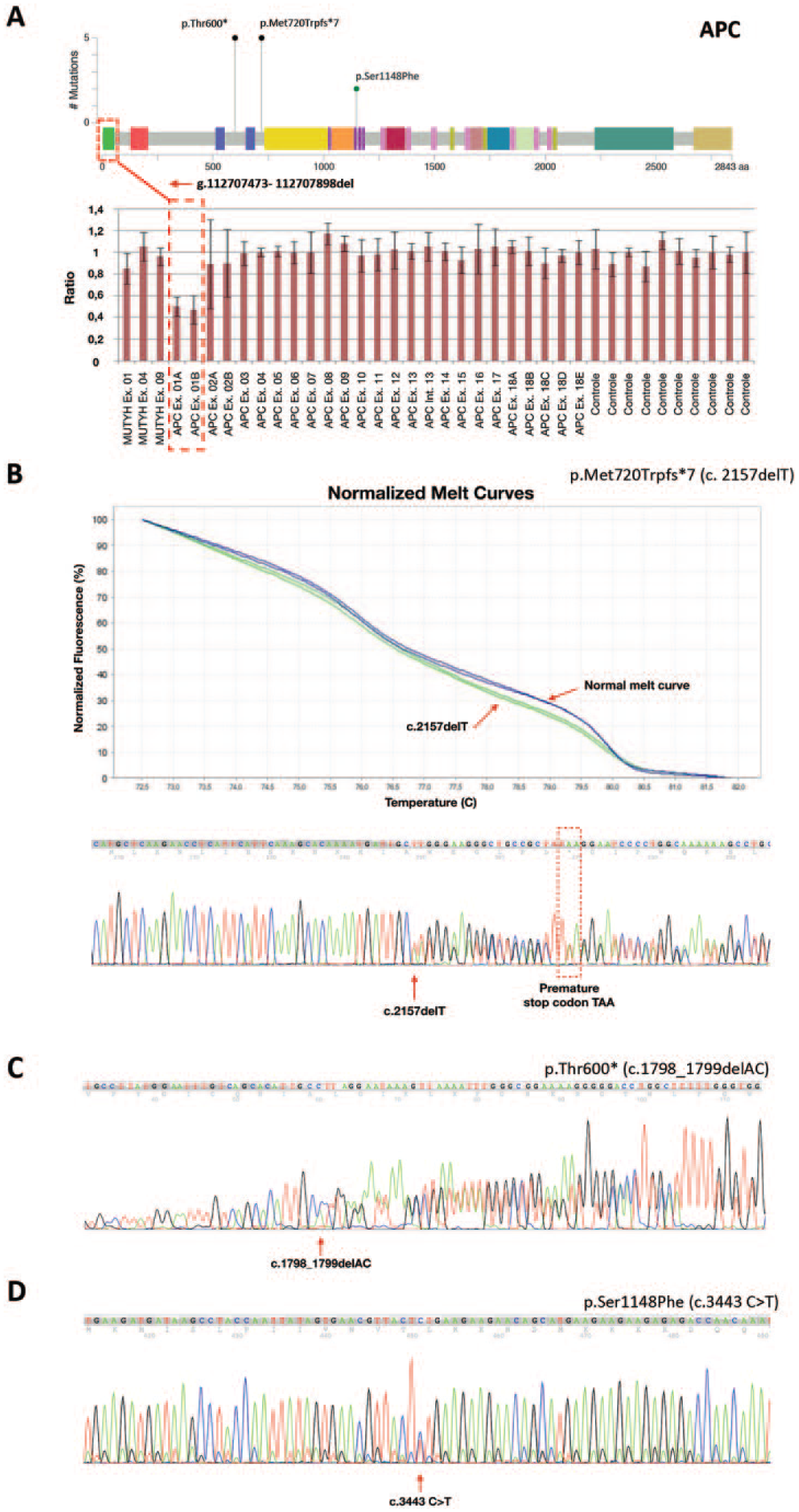
APC mutations. (a) Schematic representation of the APC gene showing four novel mutations found in this study. Black circles represent nonsense mutations; the blue circle represents a missense mutation; and the red square delimits the deleted region. The deleted region is also represented in the MLPA graph with the normalized probe rations showing the 426bp-deletion found in one family of our cohort. (b) HRM normalized melt curve is showing a blue and green curve. Curves differences implied distinct DNA sequences. In this example, the blue curve represents a non-mutated sequence, and the green curve represents the DNA sequencing harboring the novel mutation p.Met720Trpfs*7, which is further confirmed by the chromatogram below. Sequencing chromatograms show the novel mutations p.Thr600* (c) and p.Ser1148Phe (d).
MUTYH mutations
Complete MUTHY gene sequencing revealed that 30% (6/20) of the families were positive for potentially pathogenic mutations. Two missense mutations, two frameshift mutations, and one in-frame deletion were identified among the patients (Table 2). Among these mutations, two of them were novel (Figure 2(a)): p.Pro345_Pro367del (c.1029_1097del) (ID3325-Supplementary Table 1) and p.Trp349Profs*41 (c.1045_1099del) (ID3223-Supplementary Table 1). Both were found in patients diagnosed with attenuated FAP with early onset, diagnosed at ages 31 and 22, respectively. It is worth highlighting that these two deletions were found in heterozygosis and, considering the recessive inheritance of MAP, these mutations alone cannot be identified as causative of the disease. Furthermore, these two patients did not carry any other genetic variant that could be considered as compound heterozygosis along with the deletion. Likewise, the mutation p.Glu410Glyfs was also found in the heterozygosis and does not correspond to the MAP inheritance.
Two of the remaining patients carried homozygous variations (Figure 2 (b and c)), and only one individual had compound heterozygosis for two distinct missense mutations (Figure 2(d)) (ID4610-Supplementary Table 1).
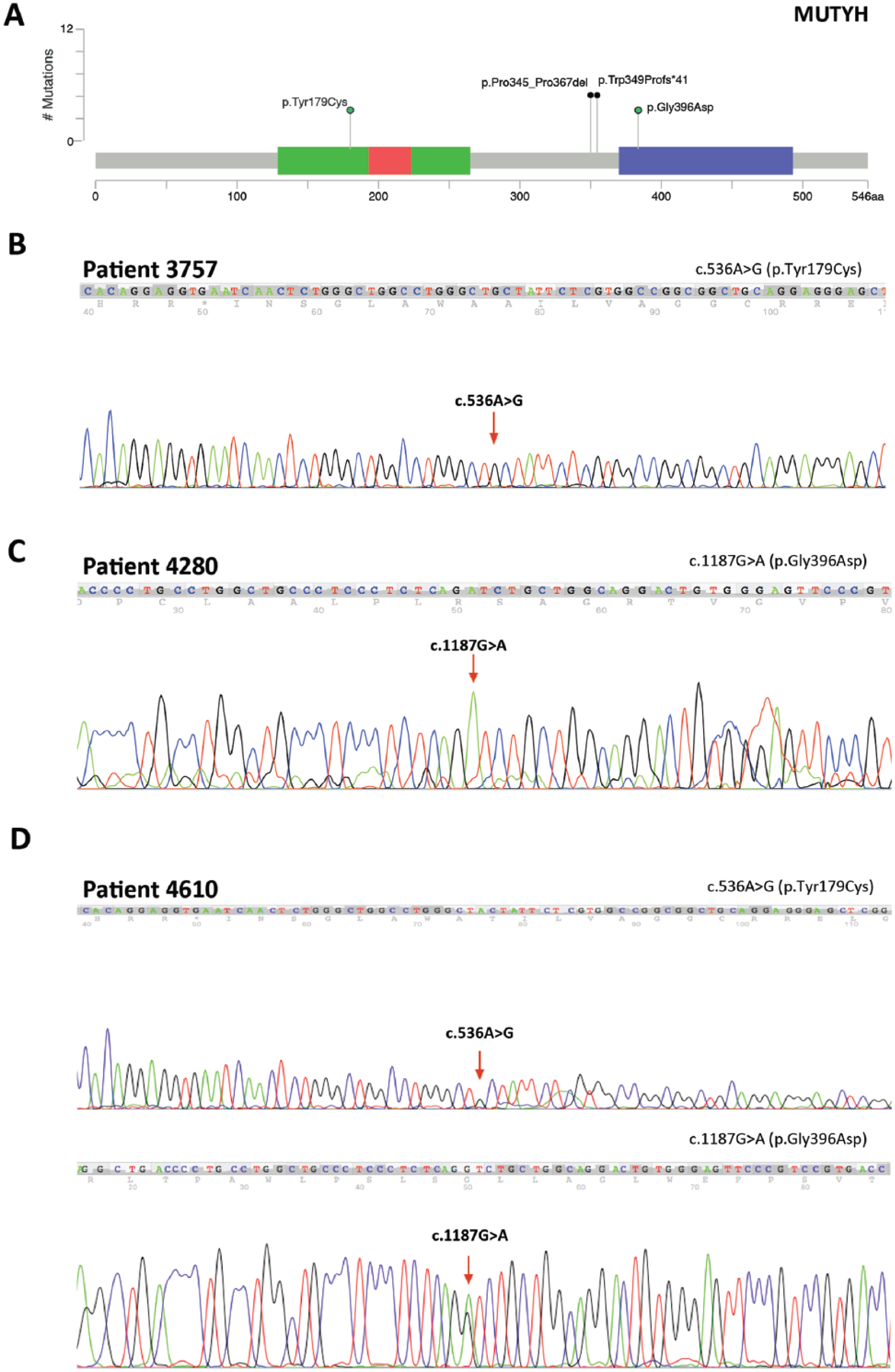
MUTYH mutations. (a) Schematic representation of the MUTYH gene showing two novel mutations found in this study (black circles) and two previously described mutations represented in the chromatogram below (green circles). Sequencing chromatograms are showing the previously described mutations p.Tyr179Cys (b), p.Gly396Asp (c), and the compound heterozygous patient, harboring both of these mutations (d).
Moreover, it is already known that large deletions are one of the mutations that lead to the loss of MUTYH gene function, and can be the causative mutations in some MAP patients. 18 Torrezan et al. 18 described a patient bearing a > 4.2-kb deletion encompassing exons 4–16 in the MUTYH gene and developed a PCR-based methodology to confirm this deletion. To investigate this large deletion, we used the PCR and MLPA approaches to screened all patients, including the three patients that carry MUTYH biallelic deleterious mutations (Supplementary Figure 1). The result of these analyses showed no large deletions in the MUTYH gene.
Genotype-phenotype correlations
Using clinical report data from the patients, we further analyzed whether the group of patients harboring APC or MUTYH mutations differ regarding clinical aspects (Supplementary Table 2). Patients carrying APC mutations had an average age of onset of 28.96 years (ranging from age 9 to 51 years), which was not statistically different from patients carrying MUTYH mutations, who had an average age of onset of 47.3 years (ranging from age 22 to 70) (Figure 3(a)) (P=0.073, Welch’s t-test). For this analysis, we removed the youngest patients from the cohort (2, 6, and 12 years old) that did not appear to have any symptoms yet. Additionally, no differences were found among the groups regarding the presence of >100 (classical) or <100 (attenuated) polyps (P= 0.131, Fisher’s exact test), a cancer familial history (P= 1.00, Fisher’s exact test), the presence of adenocarcinoma (P= 0.05, Fisher’s exact test), and extracolonic manifestations (P= 0.196, Fisher’s exact test).
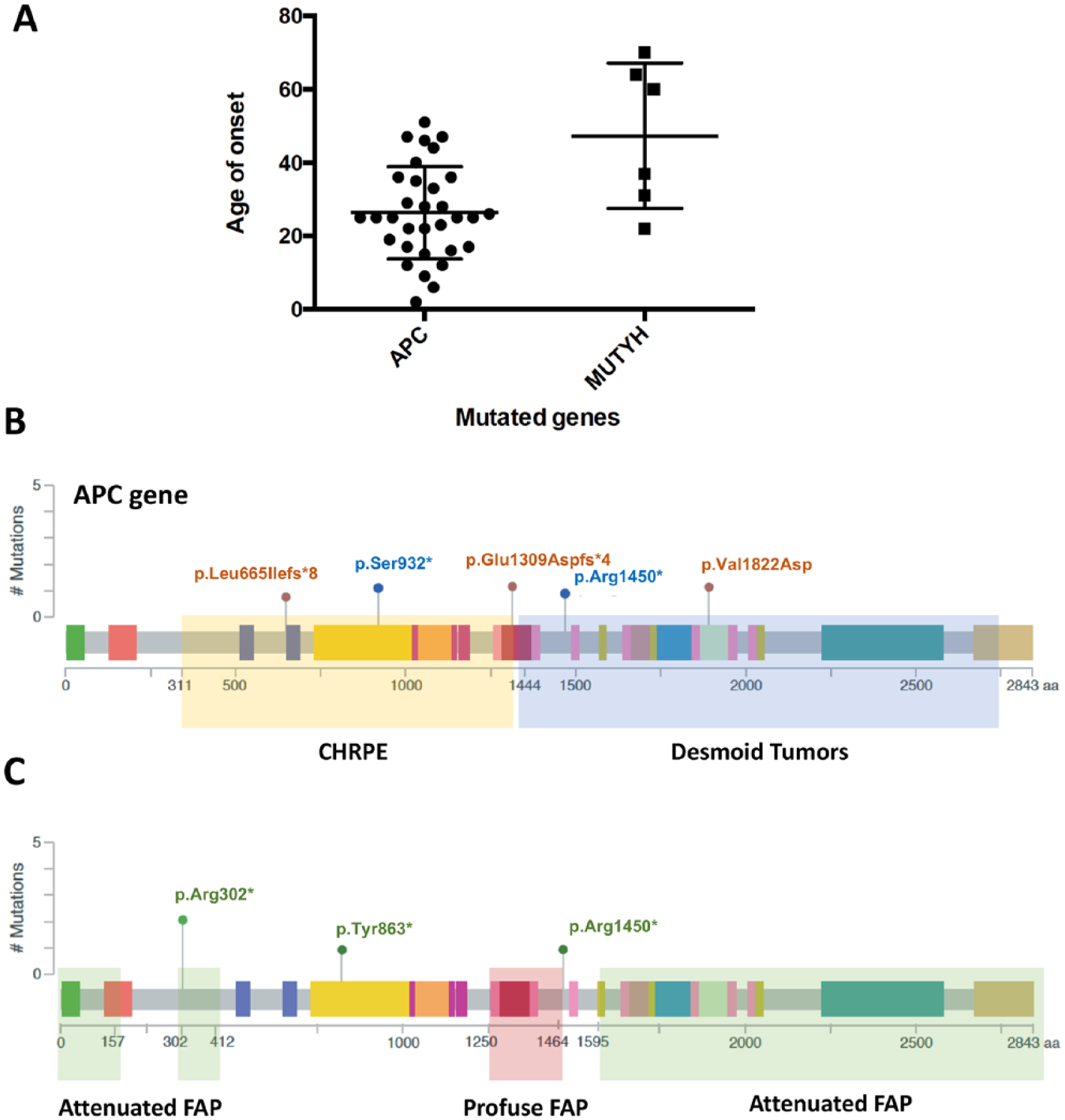
Genotype-phenotype correlations. (a) Scatter plot showing the age of onset of the APC-mutated group when compared to MUTYH-mutated patients (P=0.073, Welch’s t-test). (b) Schematic representation of the APC gene highlighting the regions previously associated with CHRPE (yellow rectangle) and desmoid tumors (blue rectangle). Brown and blue circles represent mutations that were found in patients with CHRPE and desmoid tumors, respectively. (c) Schematic representation of the APC gene highlighting the regions associated Attenuated FAP (green rectangle) and Profuse FAP (red rectangle). Green circles represent mutations that were found in APC-mutated patients attenuated FAP.
Extracolonic manifestations were observed in 50% (17/34) of the patients carrying APC mutations. Gastric polyps were the most common manifestations, occurring in 25% (10/34) of the APC-mutated patients. The second most common manifestation was CHRPE, which affected three patients, followed by desmoid tumors, which was present in two patients. Two additional APC-mutated patients had uterine dysplasia, and one patient developed osteoma. Only one MUTYH-mutated patient had extracolonic manifestations: the compound heterozygous patient (ID4610-Supplementary Table 1), who also developed gastric adenocarcinoma.
Nieuwenhuis and Vasen 19 described that mutations in specific regions of the APC gene were associated with FAP types (attenuated, intermediary and severe) and with certain extracolonic manifestations, such as CHRPE and desmoid tumors. According to the authors, CHRPE was associated with mutations between codons 311 and 1444, and desmoid tumors, with mutations beyond codon 1444 (Figure 3(b), yellow and blue rectangle, respectively). Three patients in our cohort had CHRPE: two of them indeed harbored mutations in the CHRPE-associated limit region (codons 665 and 932), but the third patient was mutated in codon 1450, a region associated with desmoid tumors (Figure 3(b), brown circles). Regarding desmoid tumors, two patients of our cohort carried this extracolonic manifestation, but only one of them was mutated within the desmoid tumor gene region (codon 1450) (Figure 3(b), blue circles).
Regarding FAP types, our cohort did not have any severe FAP patients. However, we had three APC-mutated patients diagnosed with attenuated FAP. Nieuwenhuis and Vasen 19 have described that attenuated FAP was correlated with mutations before codon 157, after codon 1595, and between codons 302 and 412 (Figure 3(c), green rectangle). Regarding the mutations found in our cohort, only one patient was mutated within the described region (Figure 3(c), green circles).
Discussion
There are extensive reports on APC and MUTYH mutational screening in FAP families; however, most of them are in European, North American, and Asiatic populations.20-22 Only one study has evaluated FAP families in the Brazilian population, 23 which has a tri-hybrid genetic background specifically, encompassing Native American, European, and African ancestries. 24 Although we have not inferred the ancestry of our cohort, De Souza et al. 25 have previously evaluated the ancestry components of patients admitted at the same hospital where we collected our samples, and showed that these patients had approximately 85% European ancestry, 10% African, and 4% Native American ancestry. These proportions are in agreement with studies that evaluated the ancestry of different cohorts, confirming the admixed unique background of the Brazilian population. 26 Mutational profiles are often population-specific; therefore, it is necessary to assess the genetic diversity and their clinical impact on different populations.
Using HRM analysis followed by direct capillary sequencing and MLPA, we evaluated 70 patients clinically suspected of FAP, and identified 16 causative mutations in 40 patients from 20 families: 16 probands were positive for APC mutations, and six probands were MUTYH mutated. Among the genetic alterations that we identified, 30% (6/20) were novel, which is a relatively high rate of novel mutation that agrees with the range of de novo alterations previously reported in different populations. 27
HRM can detect DNA sequence alteration 28 and it was chosen here as the major screening technique, since it has been reported that it has a 100% sensitivity and specificity for PCR products smaller than 300bp and can simultaneously test a large number of amplicons in a session of just a few hours.28,29 However, HRM presents some limitations that could interfere in the variant discrimination; for example, different heterozygotes may produce similar melting curves, and homozygous variant discrimination depends mostly on their melting temperature; therefore, homozygous insertion and deletions are also difficult to identify. Due to these limitations, all the samples that presented an abnormal melting curve in the HRM screening went confirmed by DNA sequencing. 30
The four new mutations identified in the APC gene were in classical FAP patients (>100 polyps), while those of the MUTYH gene were in patients with the clinical phenotype of attenuated FAP (<100 polyps), confirming that these patients were diagnosed for MAP, and not AFAP, which was the initial hypothesis. These results are in agreement with the majority of the findings regarding the genotype/phenotype correlations between APC/MUTYH mutations and FAP/MAP inheritance. 19
Three mutations in the FAP gene have already been reported in Brazilian patients. Torrezan et al. 23 found the mutations p.Arg302*, p.Arg1450*, and p.Glu1309Aspfs*4 in FAP patients with >100 polyps. In our study, two patients carried the mutation p.Arg302*, and one patient harbored p.Arg1450*. These three patients belonged to two different families (Family ID 5 and 13; Supplementary Table 1). While all family members were similar regarding the mutation, they differed in the number of polyps, suggesting a phenotypic heterogeneity among members of the same family. Additionally, the mutation p.Leu1342* was first described in the Portuguese population, specifically in an individual with early age of onset and extracolonic manifestations. Interestingly, this same mutation was found in one of our patients without any extracolonic manifestation.
Regarding the MUTYH variants, the mutations p.Pro345_Pro367del and p.Trp349Profs*41 were described for the first time in the present study. However, as they are in heterozygosis, they could not fulfill the requirements for MAP’s inheritance according to the Orphanet database. 31 The p.Tyr179Cys and mutations p.Gly396Asp, are among the most common pathogenic variants in the European population. 32 Both mutations were also previously reported in other Brazilian patients,23,33 which can be justified by the high proportion of European ancestry in the Brazilian population. 25 Additionally, Torrezan et al. 23 reported the p.Tyr179Cys mutation in patients with <100 polyps and the p.Gly396Asp in patients with >100 polyps, whereas in our studies, patients carrying these mutations presented over 100 polyps, and the compound heterozygous patient, harboring both of these mutations, had <100 polyps.
Additionally, it is worth mentioning that although we have not found an association between APC/MUTYH mutations and the presence of colorectal adenocarcinoma (Supplementary Table 2), it was previously reported that heterozygous carriers of single MUTYH mutations had an increased colorectal cancer incidence. 34 Indeed, in our cohort, among the three carriers of monoallelic MUTYH mutations, two had developed colorectal adenocarcinoma (ID1448 and ID3325; Supplementary Table 1).
Genotype-phenotype correlations have been crucial in guiding the clinical or therapeutic conduct. Earlier disease onset and polyp count information are used to distinguish the attenuated from the classical form of FAP. In this study, there was no increased risk of >100 polyps, earlier disease onset, or higher colorectal cancer risk in APC versus MUTYH patients, as should be expected.3,35 The major limitation of this study is the small number of MUTYH patients, which is possibly related to this negative result. Also, most of the patients in our cohort were initially admitted with a clinical hypothesis for classical FAP; therefore, they were first screened only for the APC gene. Thus, the attenuated FAP/MAP groups could have been underrepresented, which is implicated in the lack of association between polyp counts and APC/MUTYH mutated patients. Additionally, all the clinical data retrieved from medical records available at the Outpatient Cancer Counseling Center and information such as familial history can be highly inaccurate since they depend on the patient’s report.36,37
APC gene regions have been associated with extracolonic manifestations and polyps counts. 19 Following Nieuwenhuis and Vasen, 19 two APC-mutated patients had a mutation in the CHRPE-associated region, and one patient carried the mutation within the desmoid tumor’s region. As for polyp counts, only one patient with <100 polyps carried the mutations within the attenuated FAP-associated region. Although these mutations agree with the clinical manifestations according to Nieuwenhuis’s correlation, other patients of our cohort did not follow the same correlation. This result may be due to poor clinical information collected from these patients or be caused by other genetic factors intrinsic to these Brazilian patients.
Highlighting the phenotypic heterogeneity regarding extracolonic manifestations is critical. In our study, members of the same family developed different benign lesions. Nonetheless, the most common extracolonic manifestation found in our cohort was gastric polyps, whereas in the other Brazilian study the most frequent manifestation was desmoids tumors. 22 Moreover, from 70 patients initially screened, 42% (30/70) of them were negative for APC and MUTYH alterations. We believe that these patients must have mutations from other members of the APC and MUTHY gene pathways. Therefore, we emphasize the need for continued investigation of the mutational spectrum in distinct populations. A better understanding of the mutational spectrum of FAP patients can improve the genetic counseling of the affected families and pre-clinical diagnosis.
Conclusion
We identified 19 causative mutations in 40 Brazilian patients from 20 different families; 6 of these mutations were reported for the first time in this study. We believe that the FAP mutational spectrum can be population-specific and screening FAP patients in different populations can improve both the pre-clinical diagnosis and clinical conduct.
Supplemental Material
Supplementary_fig_1-Wilson – Supplemental material for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations
Supplemental material, Supplementary_fig_1-Wilson for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations by Luiza Ferreira Araujo, Greice Andreotti Molfetta, Otavio Costa Vincenzi, Jair Huber, Lorena Alves Teixeira, Victor Evangelista Ferraz and Wilson Araujo Silva in The International Journal of Biological Markers
Supplemental Material
Supplementary_table_2 – Supplemental material for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations
Supplemental material, Supplementary_table_2 for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations by Luiza Ferreira Araujo, Greice Andreotti Molfetta, Otavio Costa Vincenzi, Jair Huber, Lorena Alves Teixeira, Victor Evangelista Ferraz and Wilson Araujo Silva in The International Journal of Biological Markers
Supplemental Material
Supplementary_Table_V2 – Supplemental material for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations
Supplemental material, Supplementary_Table_V2 for Molecular basis of familial adenomatous polyposis in the southeast of Brazil: identification of six novel mutations by Luiza Ferreira Araujo, Greice Andreotti Molfetta, Otavio Costa Vincenzi, Jair Huber, Lorena Alves Teixeira, Victor Evangelista Ferraz and Wilson Araujo Silva in The International Journal of Biological Markers
Footnotes
Acknowledgements
We would like to thank Adriana Marques and Thais de Oliveira dos Anjos for the DNA sequencing technical support.
Author contributions
Luiza Ferreira Araujo and Greice Andreotti Molfetta equally contributed to this work.
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by São Paulo Research Foundation (FAPESP) grants #2009/53853-5, #2011/11456-0, #2013/25119-0, and #2013/08135-2; and by the National Council for Scientific and Technological Development (CNPq) grant #465539/2014-9.
Supplementary material:
Supplementary Table.xlsx – Table describing all the mutations that were found in APC and MUTYH gene.
Supplementary Table 2.doc - Genotype-Phenotype associations.
Supplementary Figure 1- Agarose gel
Manuscript word count: 3348
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Ribeirao Preto Medical School, University of São Paulo (Reference Number 3969/2012).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.