
Abstract

Foreword
Years of molecular research have described the qualitative and quantitative changes subverting cell physiology in pathological states. In cancer, accessibility to tumor biopsies has granted detailed analysis of the mechanisms leading to uncontrolled cell growth, spreading, and metastasis, allowing focused diagnosis and innovative treatments. For public health, a decisive step forward will be the availability of non-invasive tests to detect early signs of disease, establish the responsiveness to drugs, and anticipate relapses. This paradigm could be applied to any kind of human disease. A recently proposed blood test, which has wide press echoes, measures a “profile” of DNA mutations and proteins in blood and claimed a 55% success rate for early cancer detection. 1 Among novelties, the recent findings of galaxies of non-coding RNAs (ncRNAs) in human tissues and in body liquids has generated a flurry of reports proposing these molecules as biomarkers. Besides undoubted interest, we suggest that more stringent and adequate quality criteria should be established in order to consider and publish reports on ncRNAs as biomarkers.
Genomics advances: transcriptomics
After the end of the Human Genome Project in 2003, and thanks to the technological revolution represented by next generation sequencing (NGS), advancements in understanding genetic variation as well as different functional facets of genomes in normal or diseased tissues were impressive. DNA, RNA, and protein sequences from thousands of samples, either human tissues or cells and animal models, and experimental or clinical conditions, are accumulated and publicly available. RNA-seq gives access to all transcribed parts of the genome irrespective of whether they are previously known, and at unprecedented sensitivity. The conclusions reached by these “transcriptomics” studies were largely unexpected and in some sense paradoxical. Facing the known number and extension of DNA sequences encoding proteins (summing to no more that 2% of our genome), transcripts were found to cover up to two-thirds of the entire genome sequence.2,3 A second phenomenon that displayed unexpected proportions was the fact that both protein-coding and non-coding genes produce several different RNA transcripts, due to either alternative exon splicing or the use of alternative promoters or alternative polyadenylation sites.4,5 Figure 1 reports the number of RNAs currently catalogued in a database, drawing an impressive picture of an extremely active genome, where transcription is pervasive and exceedingly diversified. The old concept of one-gene-one-protein, as well as the definition of “gene” itself, needs reconsideration, 6 as illustrated in Figure 2.

The number of RNAs currently catalogued in database. The number of genes and transcripts reported in the current version of GENCODE (https://www.gencodegenes.org/). Long non-coding RNAs are molecules >200 nt with no obvious coding frame, and this number comprises intergenic, intragenic, and antisense diverging transcripts. Small non-coding RNAs comprise miRNAs (1881) and all other small RNAs (tRNAs, snoRNAs, snRNAs, and many other types). circRNA numbers are derived from the current version of http://www.circbase.org/.
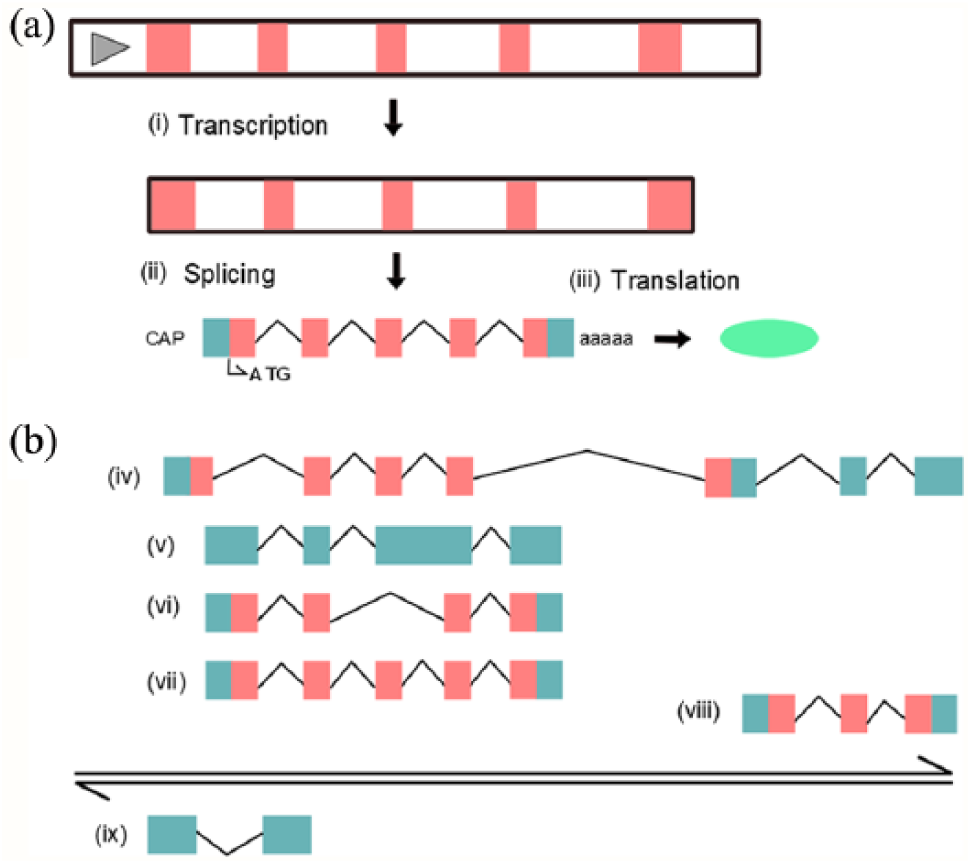
Gene and transcription-unit scheme. In (a), the classical scheme of “pre-genomic” genetic flow. In (b), a scheme featuring how genes appear based on genome browsers today. The double arrows indicate the sense and antisense strands of DNA. Above and below these arrows, sense transcripts and antisense transcripts are indicated, respectively. Colored boxes indicate exons (the parts remaining in mature RNAs), while the black connecting lines indicate introns (not in scale). Orange boxes: coding sequences. Cyan boxes: UTR and non-coding sequence. Transcript (vii) in (b) is the same as in (a) indicating the transcript in database “before” post-genomic studies. (vi) This is a second protein-coding mRNA produced by alternative splicing of the third exon. In (v) the usage of a cryptic intronic splice site has disrupted the coding frame and the RNA is entirely non-coding. In (iv) the usage of an alternative upstream promoter determines a novel first exon that, together with exons in common with (vi) and (vii) and with other distant exons farther downstream, composes a new mRNA encoding a protein with alternative N-term and C-term. In (viii), the usage of a further alternative promoter produces a coding transcript that has nothing in common with (vii) but shares the last exons with (vi), read in a different frame. On the antisense strand, a promoter overlapping the second exon of (vii) produces an antisense long noncoding RNA (ix). This scheme does not refer to any existing locus, but sums up different situations observed in many genes. (Modified from Mudge JM, Frankish A, Harrow J. Functional transcriptomics in the post-ENCODE era. Genome Res. 2013;23:1961–73. doi: 10.1101/gr.161315.113.)
Short and long
Classification as “short” or “long” noncoding RNAs is essentially technical, referring to the length in nucleotides of these molecules. Short-non-coding RNAs include micro-RNAs (miRNAs), which represent the more mature field of research, but also less studied categories such as piRNAs, endogenous siRNAs, and older entities, such as tRNAs, snoRNAs, U-RNAs, and a few others.
Long-non-coding RNAs (lncRNAs) comprise a huge number of heterogeneous RNA molecules longer than 200 nt, with no obvious open reading frame. lncRNAs derive from either known genes, being transcribed in sense or antisense, often involving introns, or divergent from the promoter region, or from intergenic regions. 7 They often present a genomic structure similar to protein-coding genes (with exons and introns), are found in cells on average in lower amounts than mRNAs, and most of them are exclusively nuclear. Functions are known for a handful of them, either as scaffolds for enzyme complexes, or as molecular bridges in chromatin structure, or as “sponges” for miRNAs, illustrating a complex post-transcriptional regulatory circuitry. 8
The transcriptional complexity has increased steadily during the last decade. Alternative splicing (AS) produces mRNA isoforms encoding for versions of proteins that are subtly different from each other; for example, by including or excluding a fragment of the coding sequence (i.e. one or more exons; Figure 2). The first described mammalian AS was the CGRP/calcitonin gene where AS produces an mRNA encoding the calcitonin hormone in parathyroid cells, and an mRNA isoform in neuronal cells encoding the neurotransmitter CGRP, 9 illustrating two fundamental properties of AS: one gene-more proteins, and tissue-specific isoforms. AS was considered anecdotal for years. Conversely, RNA-seq studies show that up to 98% of human genes undergo AS. 2 Many of these mRNA isoforms are tissue-specific; they also display specificity to different forms of disease. 10 A particular form of RNA, in which the downstream border of an exon is spliced to the preceding upstream border (rather than to the next downstream border), is represented by circular RNAs (circRNAs). 11 To detect circRNAs in RNA-seq data, special algorithms are used and validation by polymerase chain reaction (PCR) is obtained using primers designed on a reconstructed back-spliced junction (Figure 3). In each tissue, from 3 to 5000 circRNA species can be detected. Their function is still a mystery, from the re-creation of novel ORFs, 12 to functioning as miRNA sponges, 13 or to simply represent a way to reduce the production of linear mRNA. Whatever their function, splicing isoforms, including circRNAs, will greatly expand the armamentarium for investigating disease-specific RNAs.

circRNAs backsplicing. This scheme represents the internal part of a gene spanning two exons. In (a), (b), and (c), the different schemes of circularization are shown. In (a) a monoexonic circRNA is formed by backsplicing limited to exon 1. In (b) the circularization involves both exons. In (c) the same backsplicing event, but the intervening intron is retained. Small arrows indicate the primers that are used for RT-PCR analysis of circRNAs.
Exploiting potentials
The number of papers reporting specific ncRNA expression in several cancer types and other human diseases is difficult to enumerate. Many reports are based on observations conducted using model systems and then tentatively transferred to the clinical settings. Of course, these studies constitute the necessary background in the view of searching for non-invasive tests in liquid biopsies. It can be surprising that so many RNA molecules, deriving from exfoliated and dying cells, either normal or tumor,14,15 or from exosomes, 16 are found in the acellular fraction of the blood (serum or plasma) or urine, 17 since in the laboratory RNA is known as a labile molecule. In addition to being loaded into exosomes, RNA molecules are usually complexed with RNA-binding proteins that would physically protect them from degradation. It is important to emphasize that from the analytical point of view, nucleic acids are by far the most convenient kind of molecules. Thanks to PCR, the sensitivity is theoretically at the level of single molecules, and specificity is very high due to the ease of sequencing.
The most mature field is represented by miRNAs that have been extensively characterized in experimental model systems and in human tissues, and that are measurable in blood, either in serum/plasma or in isolated exosomes. 18 A number of studies have shown miRNAs as valid markers for detection, diagnosis, and prognosis of several diseases.19,20
LncRNAs possess all features of ideal biomarkers for human disease. Their expression is much more cell-, tissue-, pathology-, and stage-specific than protein coding genes. The specificity of lncRNAs expression among different human tumors is well established, also when considering tumor subtypes or drug sensitivity, as our lab and others have reported for luminal-type breast cancer.21,22 Conversely, reports on the presence of specific lncRNAs in body fluids as biomarkers are still sporadic, yet very important: the PCA3 lncRNA, the most prostate cancer-specific gene, is detectable in urine. The PROGENSA PCA3 test is the first urine RNA-based molecular diagnostic test approved in clinical routine. 23 For breast cancer, only GAS5 and H19 lncRNAs were detected in sera from breast cancer patients and were proposed as biomarkers.24,25
RNA splicing is often altered in diseases and, in cancer, mutations that either hit the splice sites or alter a splicing factor are known. 10 However, we have not found reports regarding the detection of RNA isoforms in body fluids as correlated to disease. In contrast, since 2013 the field of circRNAs has been quite productive, and several studies have described the presence of these molecules in many different tissues, including blood. 26 In many types of cancer, circRNAs have been found to be dysregulated, and in this case there are reports of tumor type specificity, which our group has recently reported for luminal-type breast cancer. 27 Indeed, due to their stability, which is conferred by the lack of free ends, circRNAs should be less prone to degradation, and thus more easily detected as circulating molecules. Although some circRNAs were found enriched in serum exosome compared to their linear mRNA counterparts, 28 robust studies on circRNAs detection in serum are still lacking.
Warning: public data digging and incongruous reporting
Due to free access to microarray and RNA-seq data in public databases, papers reporting the re-analysis of these data in search of ncRNA profiles specific to a certain type of pathology are very common today. Sometimes, findings are validated by qRT-PCR using novel sample cohorts, but often they report mere re-analysis. There is a real flurry of these types of papers, which report the association of ncRNA expression with different types of cancer and with other diseases, such as neurological and inflammatory diseases. While this work is undoubtedly important, our personal experience as readers or reviewers of a significant number of manuscripts is that many recently published (or proposed) articles concerning lncRNAs/circRNAs in clinical contexts have to be taken with care. Many studies used Gene Expression Omnibus (GEO) data irrespective of the kind of analytical platform used to generate the data. Very often, studies mix up data from multiple GEO items generated with different platforms (e.g. microarrays from Affymetrix and from Agilent) with no evident consideration of the different probes used, the different dynamic ranges, and many other sensible parameters. In a few specific cases, we have also noticed mixing together data generated by microarrays and RNA-seq that are definitely not comparable, unless thorough examination of the structure of the transcripts, the mapping of microarray probes, and the accurate analysis of raw data with adequate and robust normalization has been performed. In the case of lncRNAs, we wish to put forward another problem: some papers report results obtained on data generated by microarray analysis. Until few years ago, the most commonly used microarray platforms (e.g. the Affymetrix Human Genome U133 Plus 2.0) contained only a very limited number of lncRNA probe sets. Even in the case that some clinical correlations are found, we would suggest not spending much time considering lncRNAs that most likely are not the best obtainable using more recent technologies (such as NGS, of course). In the case of circRNAs, we have observed another kind of (worrying) problem in published studies; that is, using a backsplice searching algorithm on datasets generated by poly(A+)-RNASeq. 29 Since circRNAs do not possess poly(A)-tails, circular RNA molecules present in poly(A+)-RNA fractions would be strongly biased toward those containing short (A) stretches in exons or unspliced introns, such as Alu sequences or other repetitive elements.
A final curiosity about the geographical distribution of lncRNA papers. In PubMed searches on “cancer” and “long non-coding RNA” (and variants) limited to 2017, 55% of the records (3277/5775 on 14 February 2018) contained “China” in the affiliation. Running the same searches on a number of other terms in the second field, the percentage containing “China” was significantly lower (5%–20% of the total publications). Although a strong increase of overall scientific publications by Chinese scientists is recognized today, 30 it is hard to explain such a strong bias in lncRNA-related papers.
Conclusions: eager
We strongly believe that transcriptomics would bring impressive advancements in the field of biomarkers, based both on the fact that many transcripts have been shown to be extremely specific for tissue and pathology, and the tremendous advances in nucleic acid detection, quantitation, and sequencing, leading also to definite advantages in terms of cost. The application of transcriptomics to the field of biomarkers is at its infancy and, as always happens, it will take some time to have widespread awareness of the problems. Consequently, the fact that peer-reviewing filters are not yet completely adequate to this arising matter is absolutely admissible. We believe that all items related to the clinical, analytical, bioinformatics, and statistical facets of these studies should be thoroughly discussed in journals and meetings, and quality criteria put in place for publication and use of these data for clinical purposes.
Footnotes
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: V.M. was supported by Associazione Italiana per la Ricerca sul Cancro [AIRC Grant IG 15600 to MDB].