
Abstract
The aim of this study is to investigate the role of hydroxycarboxylic acid receptor 1 (HCAR1) in glutamate-induced excitotoxic neuronal injury. The effective and safe concentrations of an endogenous HCAR1 agonist (L-lactic acid) and an exogenous HCAR1 agonist (3,5-dihydroxybenzoic acid, 3,5-DHBA) were determined using a CCK-8 assay. An excitotoxic injury model was established by stimulating mouse hippocampal neuronal cells (HT22 cells) with 20 mM glutamate, followed by intervention with safe concentrations of L-lactic acid and 3,5-DHBA. Neuronal survival and apoptosis were assessed by NeuN immunofluorescence staining and a TUNEL assay. The effects of HCAR1 agonists on glutamate transport were evaluated using vesicular glutamate transporter 1 (VGLUT1) immunofluorescence staining and qRT‒PCR. Additionally, qRT‒PCR was performed to quantify the expression of a neuronal excitability marker gene (c-fos), inflammatory factors and hcar1. The results showed that HCAR1 agonists (L-lactic acid and 3,5-DHBA) significantly improved neuronal survival, reduced glutamate-induced apoptosis and decreased neuronal excitability (p < 0.05). The activation of HCAR1 inhibited the transport of the excitatory neurotransmitter glutamate (p < 0.05). The upregulation of HCAR1 expression led to the significant downregulation of the expression of inflammatory factors (il-1β, il-6 and tnf-α) (p < 0.05). In conclusion, HCAR1 activation effectively suppresses glutamate-induced neuronal apoptosis, reduces the uptake of excitatory neurotransmitters, and downregulates proinflammatory factor expression, thereby exerting neuroprotective effects.
Keywords
Introduction
Glutamate release, reuptake, conversion, and clearance constitute the glutamate‒glutamine cycle. Presynaptic neurons release glutamate into the synaptic cleft in a calcium-dependent manner, and glutamate subsequently binds to glutamate receptors located either postsynaptically or within the synaptic cleft to mediate excitatory neurotransmission. 1 To maintain glutamate homeostasis, the released glutamate is cleared via reuptake by excitatory amino acid transporters (EAATs) expressed on astrocytes and neurons rather than direct metabolism. Following reuptake by astrocyte-specific EAAT2, glutamate is converted to glutamine via glutamine synthetase through ammonia and ATP-dependent metabolic processes, thereby maintaining low extracellular glutamate concentrations. 2 Astrocytes then release the synthesized glutamine into the extracellular space, where it is taken up by excitatory neurons and converted back to glutamate through a process catalyzed by glutaminase.
Disruption of the glutamate‒glutamine cycle leads to excessive accumulation of glutamate in the synaptic cleft, which induces neuronal hyperexcitation and subsequent neuronal damage or death, a process that is involved in the pathogenesis of a variety of neurological disorders. 3 For example, in Alzheimer’s disease, impaired glutamate transporter function in glial cells causes the overactivation of N-methyl-D-aspartate receptors, triggering a cascade involving calcium influx and neurotoxicity. The “dying forward” hypothesis proposes that in amyotrophic lateral sclerosis, the degeneration of anterior horn neurons results from transsynaptic glutamate-mediated excitotoxic damage originating from cortical motor neurons. In epilepsy, both increased extracellular glutamate levels and decreased glutamine synthetase activity in astrocytes contribute to the generation of seizures. 4 Given its excitotoxic properties, glutamate is typically employed as an experimental stimulant to establish excitotoxic neuronal injury models in vitro, providing a reliable approach for investigating excitotoxicity-related neuropathology.5,6
Hydroxy-carboxylic acid receptor 1 (HCAR1) is a G protein-coupled receptor also known as G protein-coupled receptor 81 (GPR81). Initially, GPR81 has attracted attention because it mediates a variety of signaling functions, including the inhibition of adenylate cyclase activity, the regulation of vascular tone, and the modulation of redox homeostasis.7–9 In the past decade, GPR81 was renamed HCAR1 because it was characterized as a selective lactate sensor, demonstrating that lactate functions not only as an energy metabolite but also as a signaling molecule.10–12 L-lactic acid and 3,5-dihydroxybenzoic acid (3,5-DHBA) are ligands of HCAR1 as well as endogenous and exogenous agonists, respectively. After being bound by its ligands, HCAR1 regulates redox reactions and alleviates inflammation.13,14 In the nervous system, HCAR1 is widely and highly expressed in the cerebral cortex, hippocampus, and astrocytes.7,15 In recent years, the activation of HCAR1 has been shown to increase neurogenesis, improve synaptic plasticity, reduce neuroinflammation, and promote myelin formation.16–19 Therefore, HCAR1 represents a novel therapeutic target for neurological disorders such as Alzheimer’s disease, ischemic brain injury and retinal diseases20–22; however, its role in excitotoxic neuronal injury remains unclear.
In this study, HT22 cells were stimulated with glutamate to establish an in vitro excitotoxic injury model. L-lactic acid and 3,5-DHBA were then administered, and neuronal damage, apoptosis and inflammatory responses were explored to investigate the effects of HCAR1 activation. Our study might help elucidate whether HCAR1 is a therapeutic target for excitotoxic neuronal injury.
Materials and methods
This study was conducted from July 2024 to April 2025, and the schematic diagram is shown in Figure 1. Schematic diagram of the experiment. Control group: cells cultured in serum-free medium without any stimulation or treatment. GLU group: cells were exposed to serum-free medium supplemented with 20 mM glutamate for 24 h to induce excitotoxic injury. Lactic + GLU group: cells were cultured with serum-free medium supplemented with 20 mM glutamate and 1 mM L-lactic acid for 24 h. 3,5-DHBA + GLU group: cells were cultured with serum-free medium supplemented with 20 mM glutamate and 0.2 mM 3,5-DHBA for 24 h.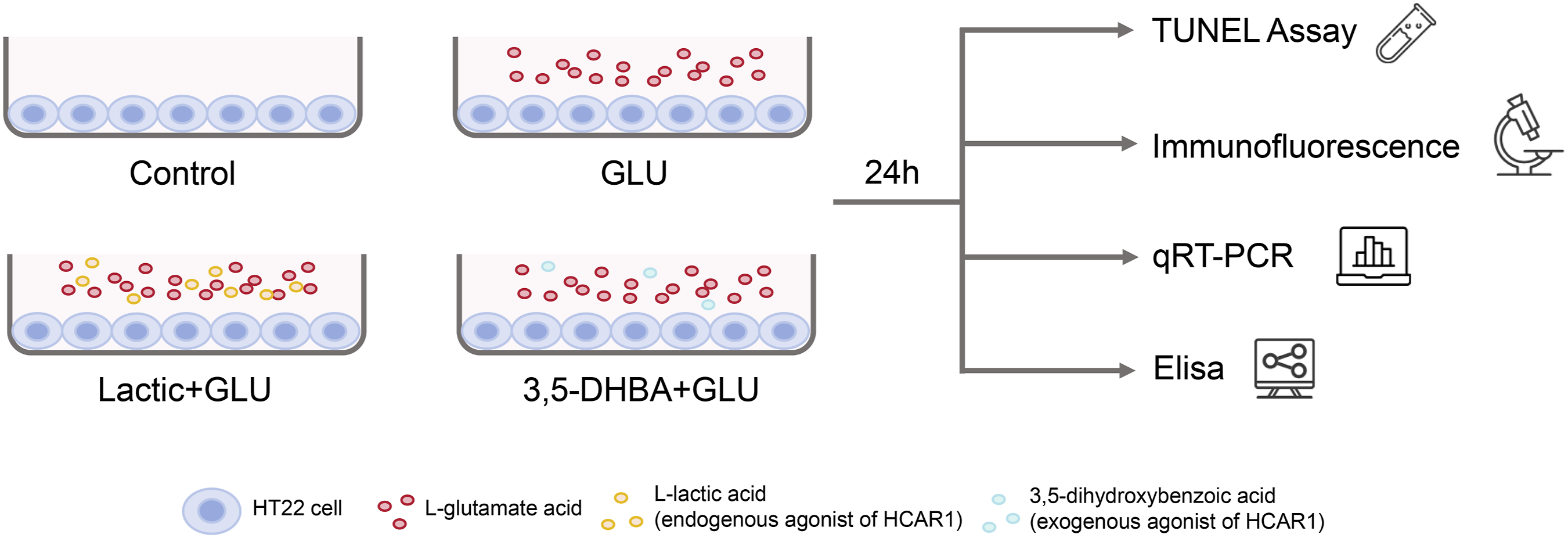
Cell line
The mouse hippocampal neuronal cell line HT22 was obtained from the American Type Culture Collection (ATCC; Mansas, VA, USA) and cultured in high-glucose Dulbecco’s modified Eagle’s medium (Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco BRL) and 1% penicillin‒streptomycin (Gibco BRL) under standard conditions (37°C, 5% CO2).
Cytotoxicity assay
The cytotoxicity of L-lactic acid (Sigma‒Aldrich, St Louis, MO, USA) and 3,5-DHBA (Sigma‒Aldrich) to HT22 cells were assessed. In brief, HT22 cells were seeded in 96-well plates at a density of 8 × 103 cells/well and incubated with culture medium supplemented with L-lactic acid (0 (control), 1, 5, 10, or 20 mM) or 3,5-DHBA (0 (control), 0.2, 0.5, 1, 5, or 10 mM) for 24 h. Cytotoxicity was assessed using a cell counting kit-8 (CCK-8) assay according to the manufacturer’s instructions (Solarbio, Beijing, China). Cell viability was calculated for three parallel replicates for each concentration to determine the concentrations of L-lactic acid and 3,5-DHBA for subsequent studies.
Excitotoxic injury induction and cell treatment
Excitotoxic injury was induced in HT22 cells with 20 mM glutamate (MedChemExpress LLC., Shanghai, China) as described previously. 23 The cells were seeded in 6-well or 24-well plates and divided into four groups (Figure 1). In the glutamate group (GLU), the cells were exposed to serum-free medium containing 20 mM glutamate for 24 h to induce excitotoxic injury. In the L-lactic acid group (Lactic+GLU), the cells were cultured with serum-free medium supplemented with 20 mM glutamate and 1 mM L-lactic acid for 24 h. In the 3,5-DHBA group (3,5-DHBA+GLU), the cells were cultured with serum-free medium supplemented with 20 mM glutamate and 0.2 mM 3,5-DHBA for 24 h. Cells cultured in serum-free medium without any stimulation or treatment for 24 h were used as the control group.
Immunofluorescence staining
HT22 cells from the four groups were seeded in 24-well plates containing slides at a density of 3 × 104 cells/well. Twenty-four hours later, the slides were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 in PBS for 20 min, and blocked with 5% normal sheep serum for 1 h at room temperature. Immunofluorescence staining was performed using a standard procedure. An anti-NeuN antibody (1:500; ab177487; Abcam, Cambridge, MA, USA) and anti-VGLUT1 antibody (1:500; SYS-135,304; Synaptic Systems, Goettingen, Germany) were used as the primary antibodies. Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:1000; Thermo Fisher Scientific, Waltham, MA) and Alexa Fluor 488-conjugated donkey anti-guinea pig IgG (1:1000; Jackson ImmunoResearch, West Grove, PA, USA) were used as the secondary antibodies. Anti-fade mounting medium supplemented with 4′,6-diamidino-2-phenylindole (DAPI; Sigma‒Aldrich) was used to label the nuclei, and after mounting, the slides were observed using a confocal fluorescence microscope (MICA; Leica, Wetzlar, Germany). HT22 cells without glutamate stimulation and L-lactic acid or 3,5-DHBA treatment were used as the control group.
TUNEL assay
HT22 cell death was evaluated using a one-step TUNEL apoptosis assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. After being mounted with anti-fade medium supplemented with DAPI (Sigma‒Aldrich), the HT22 cells on the slides were imaged using a confocal microscope (MICA, Leica). The excitation wavelengths were 488 nm and 405 nm. HT22 cells without glutamate stimulation and L-lactic acid or 3,5-DHBA treatment were used as the control group.
Quantitative real-time PCR (qRT‒PCR)
Primer sequences for qRT-PCR.

Enzyme-linked immunosorbent assay (ELISA)
Total protein was extracted from cells in the four groups and quantified. The protein levels of IL-1β, IL-6, TNF-α, and CCL2 were measured using an ELISA kit (Elabscience, Wuhan, China) according to the manufacturer’s instructions. The optical density (OD) was measured by a microplate reader (Infinite 200 PRO, Tecan Company, Switzerland) at a wavelength of 450 nm.
Statistical analysis
ImageJ software (v1.8.0; National Institutes of Health (NIH), Bethesda, MD, USA, https://rsb.info.nih.gov/ij/) was used to quantify the fluorescence intensity and area of staining in the TUNEL staining images and the NeuN and VGLUT1 immunostaining images. Statistical analysis was performed using Prism 8.0 software (GraphPad Software, La Jolla, CA, USA). Differences among the groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Statistical significance was defined as p < 0.05.
Results
HCAR1 activation improves the survival of neuronal cells
In this study, L-lactic acid and 3,5-DHBA were used to activate HCAR1 endogenously and exogenously, respectively. The cytotoxicity of the two agonists was evaluated via a CCK-8 assay. The cell viability decreased gradually as the concentration of L-lactic acid increased from 1–10 mM; however, it decreased significantly when the concentration of L-lactic acid was 20 mM (Figure 2(A); ANOVA, ****p < 0.0001). Similarly, no significant cytotoxicity was observed following treatment with 0.2–5 mM 3,5-DHBA; however, a marked decrease in cell viability was observed when the 3,5-DHBA concentration was 10 mM (Figure 2(B); ANOVA, ****p < 0.0001). Therefore, 1 mM L-lactic acid and 0.2 mM 3,5-DHBA were selected as the optimal doses for subsequent treatment experiments. An in vitro excitotoxicity model was then established by stimulating HT22 mouse hippocampal cells with 20 mM glutamate.6,23 The surviving neuronal cells were labeled with NeuN immunofluorescence staining. Following HCAR1 activation (Figure 3(A); ANOVA, ****p < 0.0001), fewer NeuN-positive cells were detected in the GLU group than in the control group (Figure 3(B)–(C); ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). However, compared with the GLU group, both HCAR1 agonist-treated groups (L-lactic acid and 3,5-DHBA groups) presented markedly greater NeuN/DAPI intensity ratios and NeuN-positive areas (Figure 3(B)–(C); ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). These findings not only demonstrate that glutamate stimulation leads to neuronal loss but also indicate that either endogenous or exogenous activation of HCAR1 improves neuronal survival under excitotoxic conditions in vitro. Viability of HT22 cells incubated with L-lactic acid and 3,5-DHBA (A) viability of HT22 cells incubated with different concentrations of L-lactic acid (0 (control), 1, 5, 10 and 20 mM) for 24 h (n = 5 for each concentration) (B) viability of HT22 cells incubated with different concentrations of 3,5-DHBA (0 (control), 0.2, 0.5, 1, 5, and 10 mM) for 24 h (n = 5 for each concentration). Differences were evaluated by one-way ANOVA followed by Tukey’s post hoc test (****p < 0.0001). Neuronal cell survival following HCAR1 activation by L-lactic acid or 3,5-DHBA treatment (A) the relative mRNA expression levels of hcar1 in HT22 cells from the control group, glutamate stimulation group (GLU), glutamate stimulation and L-lactic acid treatment group (Lactic + GLU), and glutamate stimulation and 3,5-DHBA treatment group (3,5-DHBA + GLU) (n = 4 in each group) (B and C) quantification of (B) the NeuN/DAPI intensity ratio and (C) the NeuN/DAPI area of NeuN-expressing cells in the four groups (n = 4 in each group) (D) fluorescence images of NeuN-stained HT22 cells in the four groups. Scale bar: 100 μm. Differences were evaluated by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).

HCAR1 activation reduces neuronal apoptosis and excitability
A TUNEL assay was performed to evaluate apoptosis. More apoptotic cells were detected in the GLU group than in the control group; however, the number of apoptotic cells decreased substantially following treatment with L-lactic acid or 3,5-DHBA (Figure 4(A) and (B); ANOVA, ****p < 0.0001). To assess excitability, the expression level of c-fos, a marker of neuronal activation, was analyzed via qRT‒PCR. Glutamate stimulation resulted in elevated c-fos expression in HT22 cells, whereas treatment with either L-lactic acid or 3,5-DHBA effectively reduced c-fos expression (Figure 4(C); ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). These data reveal that pharmacological activation of HCAR1 attenuates neuronal apoptosis and reduces neuronal excitability in vitro. Effects of HCAR1 activation on neuronal apoptosis and c-fos expression (A) fluorescence images of TUNEL-stained HT22 cells in the control group, glutamate stimulation group (GLU), glutamate stimulation and L-lactic acid treatment group (Lactic + GLU), and glutamate stimulation and 3,5-DHBA treatment group (3,5-DHBA + GLU). Scale bar: 100 μm (B) statistical analysis of the TUNEL-positive/DAPI-positive area ratio (n = 4 in each group) (C) the relative mRNA expression levels of c-fos in the four groups (n = 4 in each group). Differences were evaluated by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
HCAR1 activation inhibits vesicular glutamate uptake in neurons
Immunofluorescence staining of vesicular glutamate transporter 1 (VGLUT1) was conducted to investigate glutamate transport in neurons. VGLUT1-positive signals were weak in the control group; in contrast, numerous bright fluorescent signals were observed following glutamate stimulation, suggesting enhanced glutamate transport during excitotoxic injury (Figure 5(A)). However, when the HT22 cells were simultaneously treated with HCAR1 agonists (L-lactic acid or 3,5-DHBA) and glutamate, the expression of VGLUT1 significantly decreased, as indicated by decreases in both the VGLUT1 fluorescence intensity and the VGLUT1-positive area (Figure 5(A)‒(C); ANOVA, **p < 0.01, ***p < 0.001, ****p < 0.0001). To further quantify glutamate transport, vglut1 expression in the four groups was measured via qRT‒PCR. Vglut1 mRNA expression was downregulated in the two HCAR1 agonist-treated groups compared with that in the GLU group; moreover, no significant difference was found between the control group and the two HCAR1 agonist-treated groups (Figure 5(D); ANOVA, **p < 0.01, ***p < 0.001, ****p < 0.0001). Taken together, these findings indicate that in vitro pharmacological activation of HCAR1 leads to the inhibition of vesicular glutamate uptake in neurons. The expression of VGLUT1 following glutamate stimulation and HCAR1 agonist treatment (A) fluorescence images of VGLUT1 staining of HT22 cells in the control group, glutamate stimulation group (GLU), glutamate stimulation and L-lactic acid treatment group (Lactic + GLU), and glutamate stimulation and 3,5-DHBA treatment group (3,5-DHBA + GLU). Scale bar: 200 μm (B and C) quantification of the (B) VGLUT1/DAPI intensity ratio and (C) area of VGLUT1-expressing cells (n = 4 in each group) (D) the relative mRNA expression levels of vglut1 in the four groups (n = 4 in each group). Differences were evaluated by one-way ANOVA followed by Tukey’s post hoc test (**p < 0.01, ***p < 0.001, ****p < 0.0001).
HCAR1 activation suppresses proinflammatory cytokine expression
qRT‒PCR analysis was carried out to measure the expression of proinflammatory cytokines. The mRNA levels of il-1β, il-6, tnf-α, and ccl2 were markedly increased following glutamate stimulation but significantly reduced in both the L-lactic acid- and 3,5-DHBA-treated groups (Figure 6(A)‒(D); ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). The protein levels of IL-1β, IL-6, TNF-α, and CCL2 were measured via ELISA and followed the same trends as those determined by qRT‒PCR (Figure 6(E)–(H); ANOVA, **p < 0.01, ***p < 0.001, ****p < 0.0001). These results suggest that pharmacological activation of HCAR1 significantly alleviates neuroinflammation. The expression of proinflammatory factors following glutamate stimulation and HCAR1 agonist treatment (A–D) the relative mRNA expression levels of (A) il-1β, (B) il-6, (C) tnf-α, and (D) ccl2 in the control group, glutamate stimulation group (GLU), glutamate stimulation and L-lactic acid treatment group (Lactic + GLU), and glutamate stimulation and 3,5-DHBA treatment group (3,5-DHBA + GLU) (n = 3 in each group) (E–H) the protein expression levels of (E) IL-1β, (F) IL-6, (G) TNF-α, and (H) CCL2 in the four groups (n = 4 in each group). Differences were evaluated by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
Among all the subclones derived from the immortalized mouse hippocampal cell line HT-4, the HT22 cell line is the most sensitive to glutamate. 24 Owing to their ease of culture and high reproducibility, HT22 cells have become valuable models in neuroscience research, including studies on neurodegenerative injury, excitotoxicity, and neuroprotective drug screening.25–27 In this study, HT22 cells were stimulated with 20 mM glutamate for 24 h to establish an in vitro excitotoxic injury model, as verified by the high expression of the immediate-early gene c-fos. HT22 cells undergo cell death and apoptosis when large amounts of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) are released, mimicking pathological changes under excitotoxic conditions.28,29
The vesicular glutamate transporter (VGLUT) is an active transport protein in synaptic vesicles that plays a crucial role in glutamatergic neurotransmission. 30 As a member of the solute carrier 17 (SLC17) family, VGLUT comprises three main isoforms: VGLUT1 (Slc17a7), VGLUT2 (Slc17a6), and VGLUT3 (Slc17a8). 31 These isoforms are distinctly distributed in the central nervous system: VGLUT1 is predominantly expressed in the cerebral cortex, hippocampus, and cerebellum; VGLUT2 is localized mainly to the thalamus, brainstem, and spinal cord; and VGLUT3 is expressed at relatively low levels and is found in GABAergic, monoaminergic, and cholinergic neurons.32–35 These differences in regional distribution suggest that VGLUT1 is the subtype most strongly associated with neuronal excitability disorders. VGLUT1 is a glutamatergic synapse marker that is significantly upregulated in hippocampal tissues from both temporal lobe epilepsy patients and rodent models of excitotoxicity-related pathologies.36,37 Consistent with these findings, we observed a marked increase in VGLUT1 expression following glutamate stimulation in our in vitro model, reflecting increased neuronal glutamate transport capacity under excitotoxic conditions.
To date, electrophysiological approaches have been mainly utilized to investigate the role of HCAR1 in neuronal excitotoxicity. HCAR, a member of the G protein-coupled receptor family, has three subtypes, namely, HCAR1, HCAR2 and HCAR3. In this study, L-lactic acid and 3,5-DHBA were selected to specifically activate HCAR1 and exclude HCAR2- or HCAR3-mediated effects. 22 We employed an in vitro glutamate-induced excitotoxicity model to obtain novel insights into the therapeutic potential of HCAR1 activation. Both endogenous (L-lactic acid) and exogenous (3,5-DHBA) agonists effectively mitigated key pathological aspects of excitotoxicity, including neuronal apoptosis, excessive glutamate transport, and the release of proinflammatory cytokines. Notably, quantitative analysis revealed that compared with 1 mM L-lactic acid, 0.2 mM 3,5-DHBA more strongly activated HCAR1 at the transcriptional level, despite the higher concentration of L-lactic acid. These findings align with a previous report that 3,5-DHBA has a higher binding affinity for HCAR1 than L-lactic acid does. 13 Interestingly, both agonists were similarly effective at mitigating glutamate-mediated excitotoxic damage. In addition to its role as a receptor agonist, L-lactic acid acts as an important fuel source for neurons in the brain, whereas 3,5-DHBA has been characterized as a potentially neuroprotective nutritional metabolite.38,39 This dual functionality raises compelling questions about whether their nutritional benefits synergize with HCAR1 activation to protect neurons against excitotoxicity—a promising avenue for future mechanistic studies. The activation of HCAR1 has been reported to lead to a decrease in cAMP content and the release of glucose from glycogen, limiting the deleterious effect of glucose excess and mitigating neuronal damage. 40 Collectively, our findings provide evidence to support that pharmacological HCAR1 activation confers significant neuroprotection against glutamate-induced excitotoxic injury. These results not only validate the potential of HCAR1 as a promising target for neuroprotection but also provide evidence for the development of HCAR1-targeted treatment strategies for excitotoxicity-related disorders, including cerebral ischemia and epilepsy.41,42
However, this study has several limitations. First, the underlying mechanisms through which HCAR1 activation affects excitotoxic neuronal injury should be further explored. Second, the neuroprotective effects of L-lactic acid or 3,5-DHBA treatment were validated by cell experiments. We will establish an epilepsy mouse model to investigate the effects of HCAR1 agonists in future work. Finally, the synergistic effects between HCAR1 agonists and antiepileptic drugs should also be acknowledged.
Conclusion
HCAR1 activation protects neurons against glutamate-induced excitotoxicity by attenuating apoptosis, alleviating neuroinflammation, and reducing glutamate transport via VGLUT1. HCAR1 is a promising therapeutic target for excitotoxicity-associated neurological diseases, and this study provides novel insights that could inform the future development of targeted interventions.
Footnotes
Acknowledgments
We appreciate all the reviewers who participated in the review process.
Author contributions
Conceptualization, Y.L.; methodology, X.C.; formal analysis, X.C. and X.W.; investigation, X.C., and X.W.; resources, Y.L. and B.F.; writing-original draft preparation, X.C.; writing-review and editing, Y.L.; supervision, Y.L.; project administration, B.F.; funding acquisition, Y.L. All the authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Beijing Natural Science Foundation (Grant No. 7242066) and the National Natural Science Foundation of China (Grant No. 81971739).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.