
Abstract
Severe burn is a kind of traumatic injury, which can lead to serious financial burden, high morbidity and mortality following inflammatory response and complications. Microarray analysis has emerged as a popular tool for generating gene expression data and revealing the complex gene expression patterns. The Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information has become the leading public repository of microarray data. This study aimed to study the mechanisms of severe burn. Microarray of GSE37069 was downloaded from GEO including blood samples from 244 severe burn patients and blood samples from 35 healthy controls. The differentially expressed genes (DEGs) between blood samples from healthy controls and patients were identified by t-test in the LIMMA package of R. Their interaction relationships were searched using STRING online software and then protein-protein interaction (PPI) network was constructed by Cytoscape. Using DAVID online tool, underlying functions of the DEGs involved in the PPI network were analyzed by functional and pathway enrichment analyses. We screened 541 DEGs in blood samples from severe burns patients compared with blood samples from healthy controls. Meanwhile, DEGs including MMP9, TIMP1, GZMK, GZMA, and GZMB showed higher degrees in the PPI networks. Moreover, they may function in severe burn through interacting with others. According to functional enrichment analysis, these DEGs were mainly involved in BP categories associated with inflammatory responses. MMP9, TIMP1, HGF, GZMA, GZMB, and GZMK might play important roles in severe burns.
Keywords
Introduction
Severe burns can result in severe sequelae such as heterotopic ossification and significant hypermetabolism and catabolism, which are related to higher morbidity and mortality post severe burn. 1 It is reported that patients usually have hypermetabolic, debilitated, and disabled diseases for at least 2 years after severe burn injury.2,3
After burns, inflammatory responses start immediately and are usually sustained for several months or years. 4 The inflammatory responses include many aspects such as sepsis, which is a type of systemic inflammatory response syndrome induced by infection after severe burn. 5 Moreover, the level of serum procalcitonin indicating severity of septic complication in patients with severe burn6–8 and the continuous downregulation of monocyte human leukocyte antigen-DR have correlation with septic complications.9–11 Insulin-like growth factor 1 (IGF-I)/insulin-like growth factor binding protein-3 (IGFBP-3) can reduce small bowel epithelial cell apoptosis by downregulating the Fas pathway, and then enhance gut mucosal integrity following severe burn. 12 As previously reported, tumor necrosis factor (TNF) can decrease lymphocyte apoptosis thus helping to enhance immunity and reduce risk of sepsis; 13 the elevation of MMPs may be beneficial to tissue fibrosis and scar formation after burn injury.14,15 Thus, the prognostic importance of MMPs and TIMP1 for burn trauma has been established. 16 Some of these results have been successively testified in animal models for the drug candidates for burn trauma. However, there was poor correlation in the responses between animal models and human conditions.
In 2013, Seok et al. performed a microarray data analysis based on GSE37069 (burns), GSE36809 (tranma), endotoxemia (GSE3284), and four microarray data of sepsis (GSE13094, GSE9960, GSE13015, and GSE28750) to identify gene expression changes responding to burns, trauma, sepsis, and endotoxemia between patients/mouse models and healthy controls. 17 They obtained numerous differentially expressed genes (DEGs) between burns patients and controls, and found that the DEGs and their related function terms were different between mouse models and human conditions. This demonstrated there still existed distance between clinical application and the laboratory test, and more tests should be done to identify more candidates for therapy for burn trauma.
In order to identify more genetic candidates for therapy for burn trauma, expression microarray data GSE37069 deposited in Gene Expression Omnibus (GEO) by Seok et al. 17 was downloaded. DEGs in this dataset were identified and their interaction relationships were investigated by protein-protein interaction (PPI) network. Moreover, potential functions of DEGs involved in the PPI network were analyzed by functional and pathway enrichment analyses. The functions of the significant DEGs involved in significant function terms would be discussed and some of them might be considered as the candidates for severe burn therapy.
Materials and methods
Microarray data
Expression profile of GSE37069 deposited by Seok et al. 17 which was based on the platform of GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array was obtained from GEO (http://www.ncbi.nlm.nih.gov/geo/). GSE37069 included 281 blood samples from 244 severe burn patients admitted within 96 h after burn injury and with over 20% of the total body surface area of burns, and 37 blood samples from 35 healthy controls (age range, 16–55 years; seven controls were studied twice approximately 2 years apart). The informed consents were obtained from all the participants in the study of Seok et al. 17
DEGs screening
Microarray data were preprocessed by Affy package. 18 After background correction and quantile normalization, probe IDs were converted to gene symbols and then duplicates were removed. Finally, the gene expression matrix of the test group was obtained. The t-test method in the LIMMA (Linear Models for Microarray Data) package of R 19 (http://www.bioconductor.org) was used to identify the genes that were differentially expressed between blood samples from patients and healthy controls. Next, the t-test P value was adjusted into false discovery rate (FDR) using multiple Benjamini–Hochberg tests. 20 Then, the genes of FDR <0.05 and |log2fold change (FC)| >1.2 were considered as significant DEGs in this study.
PPI network construction
The interactions of proteins encoded by the DEGs selected above were searched by STRING online software, 21 with combined score (required confidence) >0.4 was used as the cutoff criterion. Afterwards, the PPI network was visualized by using Cytoscape software (http://www.cytoscape.org). 22 Then, connectivity degree analysis in the PPI network was performed to identify the potential key nodes (proteins) in the network.
Functional and pathway enrichment analysis
Gene Ontology (GO) employs a series of structured vocabularies for annotating genes and their products in biological process (BP), cellular component (CC), and molecular function (MF). 23 Kyoto Encyclopedia of Genes and Genomes (KEGG) database is composed of known genes and their functions and informs people of how molecules or genes work. 24 Database for Annotation, Visualization, and Integrated Discovery (DAVID) is a valuable method to classify genes and terms into several manageable modules. 25 Using the DAVID online tool, we performed GO functional and KEGG pathway enrichment analyses to identify the GO BP terms and pathways associated with the DEGs involved in the PPI network. The terms and pathways connected to at least three genes at the level of P value <0.05 were considered significant.
Results
DEGs analysis
After the data processing, a total of 541 DEGs were identified between blood samples from healthy controls and patients with the FDR <0.05 and |log2FC| >1.2. Among these 541 genes which were significantly altered in burn patients, 305 genes were upregulated genes (e.g. CTSG, MPO, CAMP, PROK2, TSPO, TIMP1, LTF, HGF, STAT5B, and MMP9) and 236 were downregulated genes (e.g. CD2, CD8A, EOMES, GPR183, GZMK, GZMA, and GZMB).
PPI network analysis
We then separately searched the PPI networks of the up- and downregulated DEGs products. The PPI network of upregulated genes productions consisted of 170 nodes interacting through 384 interactions (lines) (Figure 1). Twenty-eight (16.5%, 22/170) nodes interacting with others through >8 interactions, such as translocator protein (18 kDa) (TSPO, degree = 42), myeloperoxidase (MPO, degree = 22), cathepsin G (CTSG, degree = 20), lactotransferrin (LTF, degree = 18), cathelicidin antimicrobial peptide (CAMP, degree = 16), matrix metalloproteinase 9 (MMP9, degree = 17), tissue inhibitor of metalloproteinase 1 (TIMP1, degree = 13), prokineticin 2 peptide (PROK2, degree = 8), and hepatocyte growth factor (HGF, degree = 8) which interacted with each other.

The protein-protein interaction (PPI) network for upregulated genes.
The PPI network of productions of the downregulated genes consisted of 286 interactions linking to 101 nodes (Figure 2). This PPI network contained 33 nodes (32.7%, 33/101) interacting with the others through >8 interactions, such as CD2 (degree = 28), CD8A (degree = 26), granzyme K (GZMK, degree = 23), granzyme A (GZMA, degree = 22), granzyme B (GZMB, degree = 17), eomesodermin (EOMES, degree = 8), and G-protein-coupled receptor (GPR)183 (GPR183, degree = 8).

The protein-protein interaction (PPI) network for downregulated genes.
Functional and pathway enrichment analysis
After the GO enrichment analysis, the DEGs in the PPI network of upregulated genes were mainly associated with the GO BP categories of defense response (including STAT5B, MPO, CTSG, LTF, CAMP, and PROK2, P value = 2.41E-15), inflammatory response (including STAT5B and PROK2,P value = 2.58E-09), and response to wounding (including STAT5B and PROK2, P value = 1.48E-08) (Table 1). Moreover, these DEGs were enriched in pathways such as hematopoietic cell lineage (P value = 1.04E-02), Fc epsilon RI signaling pathway (P value = 3.24E-02), and glutathione metabolism (P value = 4.20E-02) (Table 2).
The top 10 enriched Gene Ontology biological process terms for the up- and downregulated genes involved in PPI networks.

The enriched KEGG pathways for the up- and downregulated genes involved in PPI networks.
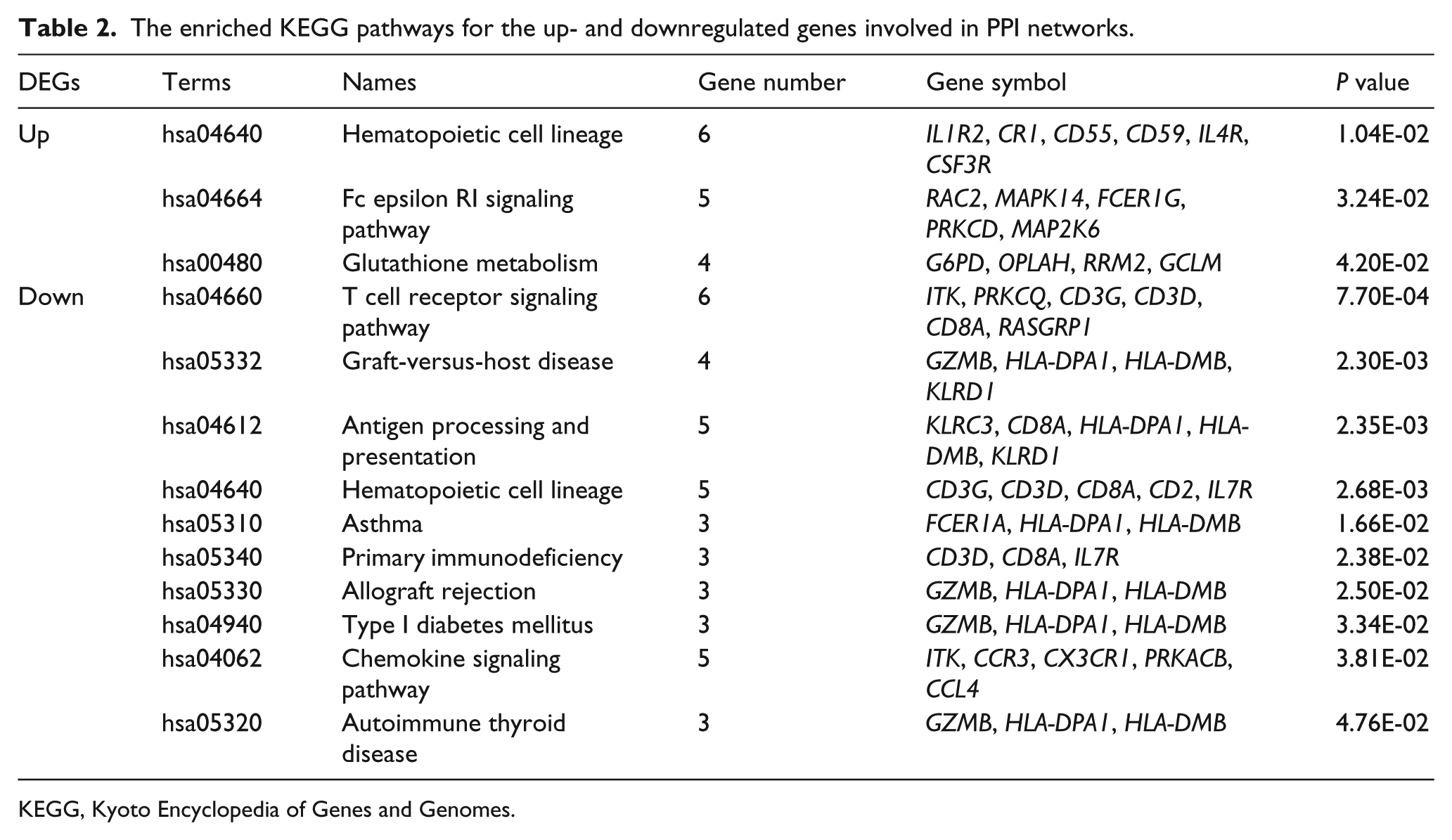
KEGG, Kyoto Encyclopedia of Genes and Genomes.
Furthermore, the DEGs in the PPI network of downregulated genes were essentially involved in the GO BP terms of T cell activation (including EOMES, GPR183, CD2, and CD8A, P value = 1.20E-06), lymphocyte activation (including EOMES, GPR183, CD2, and CD8A, P value = 4.22E-06), and immune system development (including EOMES, GPR183, and CD8A, P value = 1.73E-03 (Table 1). These DEGs in the PPI network of downregulated genes were enriched in the KEGG pathways such as T cell receptor signaling pathway (including CD8A, P value = 7.70E-04), Graft-versus-host disease (including GZMA, P value = 2.30E-03), antigen processing and presentation (including CD8A, P value = 2.35E-03), and allograft rejection (including GZMB) (Table 2).
Discussion
Using common bioinformatics methods, 26 the genes of FDR <0.05 and |log2FC| >1.2 were identified as significant DEGs, which were further screened using PPI network and enrichment analysis. In this study, a total of 541 DEGs, including 305 upregulated and 236 downregulated genes, were identified from the blood samples of severe burns patients admitted within 96 h after burn injury compared with healthy controls. These 305 upregulated genes (including STAT5B, MPO, CTSG, LTF, CAMP, and PROK2) were mainly involved in GO BP categories of defense response, inflammatory response, and response to wounding. These 236 downregulated genes (including GZMA, EOMES, GPR183, CD2, and CD8A) were essentially enriched in T cell activation, immune system development, and allograft rejection. These results indicated that those DEGs identified in this study were associated with the inflammatory response and the defense response after burn injury. And abnormal gene expressions occurred in severe burns patients admitted within 96 h after burn injury.
The dysregulation of the expressions of MMPs and their inhibitors such as TIMP1 are confirmed in diseases generating systemic inflammatory response syndrome including burn trauma. 16 Ulrich et al. 14 confirmed that the expression levels of MMP9 and TIMP1 showed a significant elevation only between 3 and 4 days post surgery. Moreover, the overexpression of MMP9 is demonstrated to be correlated with enhanced blood–brain barrier (BBB) permeability after traumatic brain injury, 27 and the increasing expression of MMPs and TIMP1 levels may be beneficial to tissue fibrosis and scar formation after burn injury.14,15 Thus, the prognostic importance of MMPs and TIMP1 for burn trauma has been established. 16 These results are inconsistent with the result in this study that the expression levels of MMP8, MMP9, and TIMP1 were upregulated in the burn patients compared with the healthy controls, demonstrating the prognostic importance of MMP8, MMP9, and TIMP1 in burn injury.
Among the DEGs after burn, genes such as STAT5B, MPO, CTSG, LTF, CAMP, and PROK2 showed a significant elevation change. All these genes are anti-inflammatory mediators. As has been shown, the constitutive activation of STAT5B is necessary for the development of Foxp3-positive Treg cell by binding directly to Foxp3 gene and enforcing it expression, which is associated with suppression of the inflammatory response. 28 MPO is a myeloid enzyme present in the polymorphonuclear leukemic cells, 29 and the synthesis of MPO is restricted to promyelocytes and selectively expressed during hemopoiesis in the granulocytic lineage. 30 Moreover, the expression of CAMP, a direct target of the vitamin D receptor encoding antimicrobial peptides, is strongly upregulated in myeloid cells after injury, revealing a importance of CAMP in the response to wounding.31,32 In addition, the CTSG gene is a serine protease expressed in promyelocytes, which might influence the risk and mortality of severe sepsis in children. 33 The expression levels of all these genes have been determined by upregulation in patients after burn injury or in diseases with systemic inflammatory response via acting directly as anti-inflammatory mediators or showing anti-inflammatory activation.34–36 Thus, the overexpression of these genes of STAT5B, MPO, CTSG, LTF, CAMP, and PROK2 revealed the anti-inflammatory responses had been greatly emerged in burn patients, and thus might be used as potential mediators to recovery from injuries.
Furthermore, as a serine protease produced by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, GZMA has correlation with the progression of sepsis in burn patients. 37 The cytolytic effector molecules GZMA and GZMB are reported to be upregulated in CTLs which are activated in severe sepsis, and their overexpression is associated with severity of sepsis. 38 In sepsis patients, GZMB-mediated lymphotoxicity results from megakaryocytes-platelet transcriptional alteration, thus GZMB can be served as a promising target for therapy of sepsis. 39 The increased expression level of GZMB has been observed during prolonged inflammation, and the upregulation of it has been implicated in several chronic inflammatory diseases. Additionally, the GZMB proteins can degrade extracellular matrix proteins including fibronectin and MMPs. 40 Moreover, the plasma GZMK levels which allows for a detailed classification of sepsis and more specific treatments can be used as a valuable marker to stage sepsis. 41 In this study, however, the expression level of GZMA, GZMB, and GZMK were downregulated in burn patients, indicating that the sepsis of these patients might not be serious and that the severe burn patients had no malignant diseases with chronic and prolonged inflammatory diseases.
Other downregulated genes such as EOMES, GPR183, CD2, and CD8A were identified in this study. GPR183 also known as Epstein–Barr virus-induced gene 2 (EBI2), controls B lymphocyte migration and is expressed in macrophages and B cells. 42 GPR183 plays an important role in the development of T-dependent early antibody responses by collaborating with the chemokine receptors such as CXCR5 to position B cells. 43 The CD8 molecules function as important co-receptors for class I major histocompatability complex on the surface of T lymphocytes, 44 suggesting the vital roles of them in antibody responses. The downregulation of these genes in burn patients in this study might suggest that the patients developed no antibody responses.
Conclusions
In conclusion, an integrated bioinformatics analysis has been conducted to identify genes which may be implicated in severe burn injuries in this study. We screened 541 DEGs between blood samples from severe burns patients and healthy controls. The results showed that, DEGs such as MMP9, TIMP1, PROK2, GZMA, GZMB, GZMK, and GPR183 might correlate with severe burns by being involved in the inflammatory responses. Nevertheless, further investigations are still necessary to verify the hypotheses proposed in this study and to unravel their mechanisms in severe burn injuries.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.