
Abstract
Objectives
To evaluate the novel Phoenix Atherectomy System as percutaneous treatment of de novo and restenotic infrainguinal arterial lesions.
Methods
This prospective, multicenter, nonrandomized investigational device exemption trial was conducted across 16 US and German centers between August 2010 and April 2013. Intention-to-treat enrollment was 128 patients (mean age: 71.8 years, 59% male) with 149 lesions (mean length: 34 mm, mean diameter stenosis: 89.5%), and the primary analysis per-protocol population consisted of 105 patients with 123 lesions. The primary efficacy endpoint, technical success, was the achievement of acute debulking with a post-atherectomy residual diameter stenosis ≤50% (before adjunctive therapy). The primary safety endpoint was the major adverse event (MAE) rate through 30 days.
Results
For the primary analysis per-protocol population, the rate of lesion technical success was 95.1% (117/123), with the lower limit of the 95% CI 90.6%, meeting the prospectively established target performance goal of ≥86%. After post-atherectomy adjunctive therapy, residual stenosis was ≤30% for 99.2% (122/123) of lesions (mean final diameter stenosis 10.5%). Improvement of ≥1 Rutherford class occurred for 74.5% of patients through 30 days and for 80% through six months. MAEs were experienced by 5.7% (6/105) of patients through 30 days (with the upper limit of the 95% CI 11.0%, meeting the target performance goal of <20%), and 16.8% through six months. Six-month freedom from TLR and TVR was 88.0% and 86.1%, respectively.
Conclusions
Based on the high rate of technical success and the low rates of MAEs through six months, the Phoenix Atherectomy System is safe and effective for the debulking of lower-extremity arterial lesions.
ClinicalTrials.gov identifier NCT01541774
Keywords
Introduction
Although endovascular intervention has overtaken bypass surgery as the more frequent, less invasive first-line approach for restoring blood flow to the lower extremities of patients with peripheral arterial disease (PAD), the optimal percutaneous technique is still not well established.1–6 Endovascular options include percutaneous transluminal angioplasty (PTA), self-expanding nitinol stents, 7 drug-eluting stents 8 and balloons, 9 and atherectomy catheters.10–17
Designed to debulk the burden of atherosclerotic plaque by increasing luminal gain with catheter-deliverable devices, atherectomy offers the theoretical advantage of overcoming the limitations of PTA, such as acute dissection and elastic recoil, thus potentially reducing the need for adjunctive stenting and the rate of restenosis.10,18–20 The currently available atherectomy modalities – designated as directional, rotational, orbital, and ablative – provide distinct approaches to the debulking and removal of plaque.10,18 Each modality has shown good technical success in procedural reduction of stenosis, effectiveness in reducing PAD symptoms, and acceptable but varying levels of adverse events including the need for revascularization procedures.13,14,17 However, significant risks remain associated with atherectomy of the superficial femoral, popliteal, anterior tibial, posterior tibial, and peroneal arteries – including arterial dissection, perforation, embolism and/or thrombosis, and restenosis. 18 Distal embolization, in particular, remains a concern – with recent atherectomy trials reporting rates in the range of 2% to 4%13,17 – warranting the employment of embolic protection devices in some circumstances.21,22
The novel Phoenix Atherectomy System (Volcano Corporation, San Diego, California) is an over-the-wire device with a front-cutting metal element at the distal tip of the catheter to treat diseased femoropopliteal and below-the-knee (BTK) arterial segments with a range of diameters, including 1.8-mm, 2.2-mm, and 2.4-mm sizes (Figure 1). The 2.4-mm device includes a deflecting mechanism, which allows debulking of arterial diameters that are larger than the catheter’s diameter. The Phoenix was designed with the intention of reducing the risk of distal embolization and negative vessel interaction, of obviating the need for removal of the device from the body during the procedure to purge collected debris, and of avoiding adverse effects (such as excessive blood removal and vessel suck-down) related to the use of aspiration.

The Phoenix Atherectomy System (Volcano Corporation, San Diego, California). (a) Catheter handle with self-contained battery-powered motor and flexible catheter shaft with minimum working length of 130 cm. (b) The inward-cutting helical blade sitting with a housing that acts as a shield.
We present 30-day and six-month outcomes from the prospective multicenter independently adjudicated Endovascular Atherectomy Safety and Effectiveness (EASE) trial, an investigational device exemption (IDE) study conducted to evaluate the technical success and safety profile of the Phoenix in a range of above-the-knee (ATK) and BTK arterial locations and diameters, PAD categories, and disease morphologies. Data analysis in the regulatory trial included comparison with prespecified target performance goals based on pivotal trial outcomes for other atherectomy devices.
Methods
Trial design
The prospective, multicenter, nonrandomized, single-arm EASE trial (ClinicalTrials.gov identifier NCT01541774) was conducted to evaluate the safety, effectiveness, and performance of the Phoenix Atherectomy System in the percutaneous treatment of de novo and restenotic atherosclerotic lesions of infrainguinal lower extremity arteries. Fourteen centers in the United States and two centers in Germany participated after receiving approval from their independent, local medical ethics committees or institutional review boards, and informed written consent was obtained from all enrolled patients. The devices, site training methods, subject eligibility, adverse event adjudication, and endpoint monitoring were identical for the US and German study sites.
Patients ≥18 years of age were eligible for the study if they had resting ankle-brachial index (ABI) ≤0.90, or ABI ≤0.75 after exercise, or noncompressible toe-brachial index (TBI) ≤0.80, and PAD assessed as Rutherford class 2 to 5. 23 Inclusion and exclusion criteria for the trial are summarized in Table 1. The trial was conducted under IDE G090095, in compliance with national and local regulations (US CFR 21 Parts 11, 50, 54, 56, and 812; ISO 14155; and EC Directive 2007/47/EC for Germany) and in accordance with the ethical principles of the Declaration of Helsinki.
Selected eligibility criteria of the EASE trial.

Patient cohort
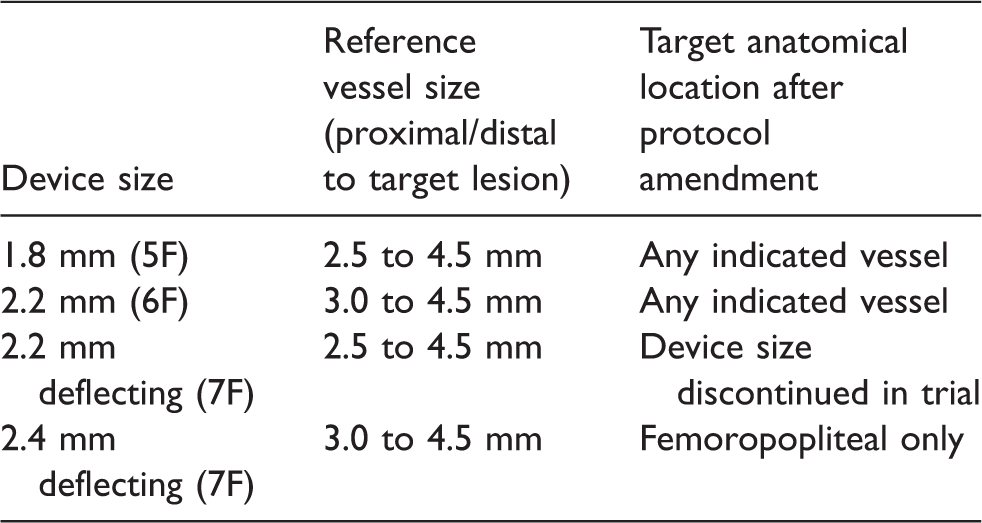
Between August 2010 and April 2013, 128 patients were enrolled in the intention-to-treat (ITT) population, 88 (69%) at the US sites and 40 (31%) at the German sites, and a total of 149 lesions were treated with the Phoenix Atherectomy System. After 36 initial patients had been treated with 2.2-mm or 2.4-mm 7F devices with deflecting catheters, the study sponsor placed a hold on enrollment pending a review to assess the underlying cause of a higher than anticipated rate of dissections and perforations: 19 serious procedure- or device-related adverse events, six of which qualified as major adverse events (MAEs) – five dissections/perforations and one vessel occlusion requiring treatment. Following review of clinical data and angiographic images by an independent imaging core laboratory service, it was determined that all six MAEs occurred below the popliteal artery and resulted from use of the 7F deflecting catheter in vessels that averaged 1.89 ± 0.66 mm in diameter. Based on these findings, in conjunction with the US Food and Drug Administration (FDA) IDE supplement process, two devices with smaller diameters (1.8-mm and 2.2-mm non-deflecting catheters) were added to the trial and a vessel-sizing algorithm was introduced (Table 2), reflecting the intended product labeling.
Protocol-defined device/vessel sizing.
Of the initial 36 patients, 23 (64%) did not meet the amended eligibility criteria, and these patients were excluded from the primary outcome analyses in the per-protocol (PP) population (while being retained in the ITT population). In this report, the PP group (105 patients with 123 lesions) is the focus of the primary analysis comparison, as these clinical results most accurately reflect the intended use (anatomic location and vessel size requirements) expressed in the device labeling. The ITT-only population of 23 patients was analyzed as a subgroup. The demographics, medical history, and risk factors for the PP and ITT populations are summarized in Table 3, and the baseline clinical assessments for the PP and ITT populations are summarized in Table 4. There were no remarkable differences between the PP and ITT populations in terms of demographics, medical history and risk factors, or baseline clinical assessments.
Demographics, medical history, and risk factors.
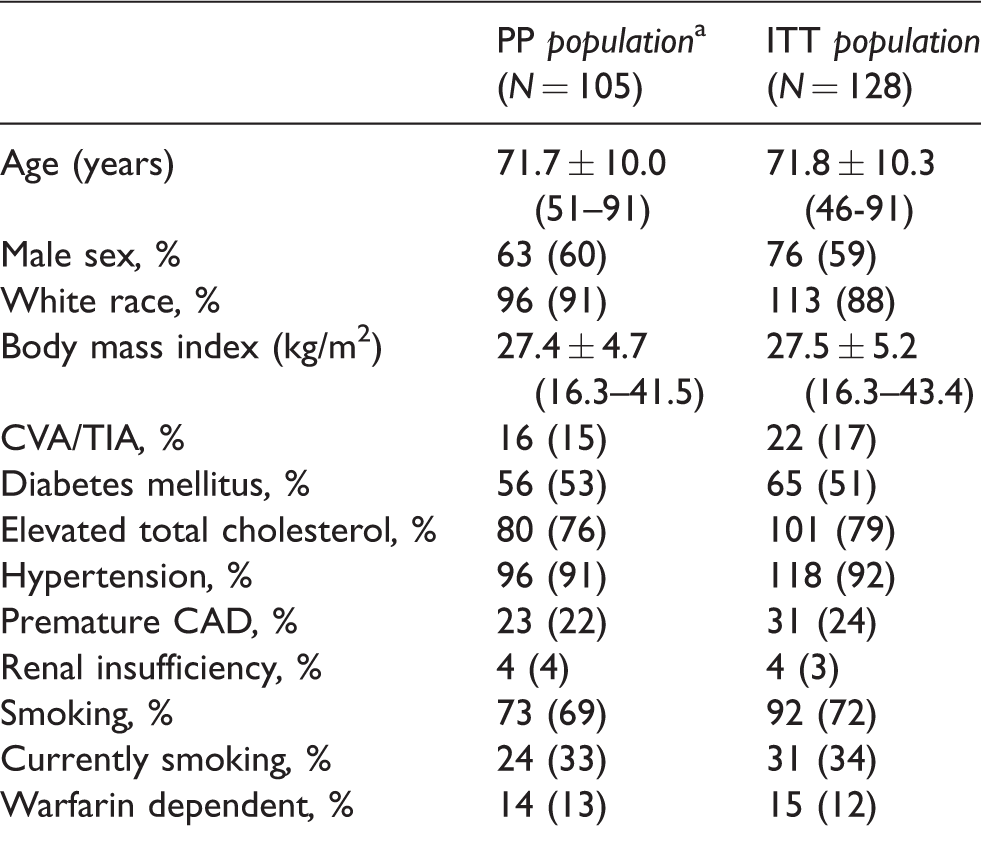
CAD: coronary artery disease; CVA: cerebrovascular accident; ITT: intention-to-treat; PP: per protocol; TIA: transient ischemic attack.
aThe original ITT population consisted of 128 patients; but after the first 36 patients had been treated, the protocol guidelines for artery diameter and device catheter sizing relative to anatomical location were amended, and as a result 23 patients were excluded from the primary outcome analyses in the per protocol (PP) population.
Continuous data are presented as the means ± standard deviation (range); categorical data are given as the counts (percentage).
Baseline clinical assessments.
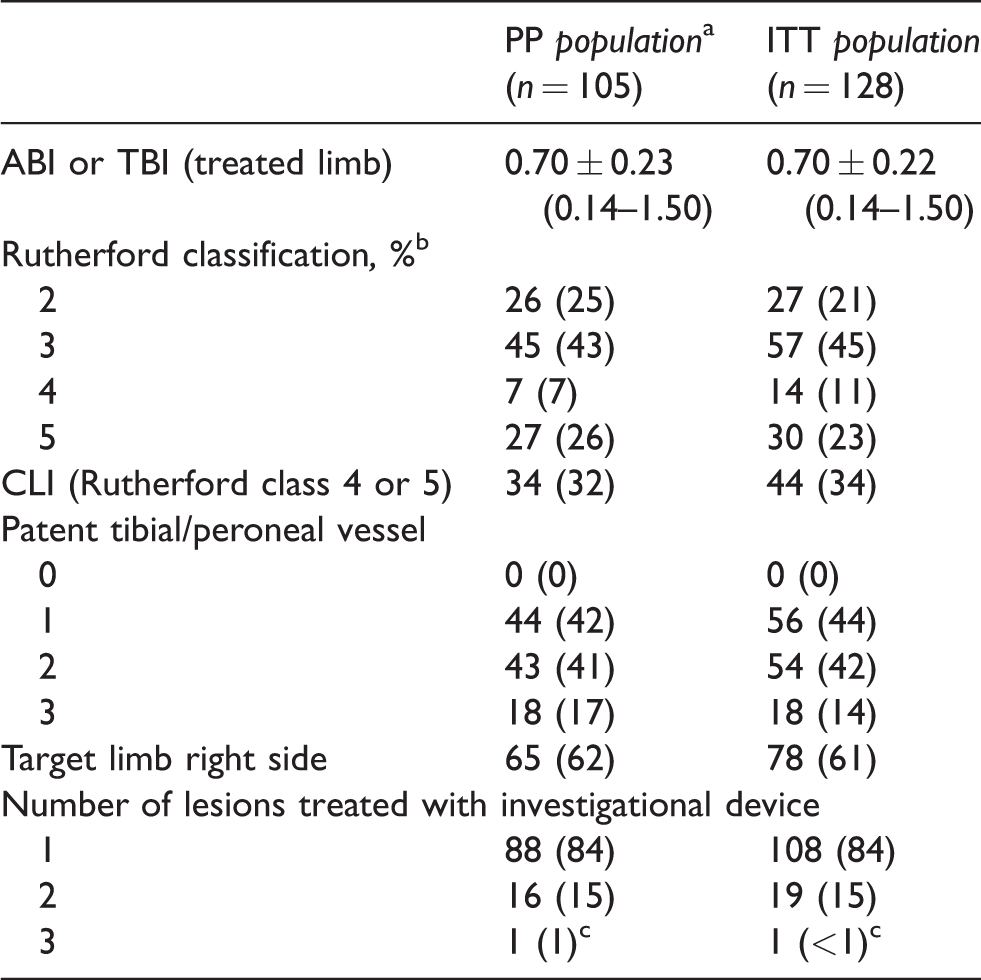
ABI: ankle-brachial index; CLI: critical limb ischemia; ITT: intention-to-treat; PP: per protocol; TBI: toe-brachial index.
aThe original ITT population consisted of 128 patients; but after the first 36 patients had been treated, the protocol guidelines for artery diameter and device catheter sizing relative to anatomical location were amended, and as a result 23 patients were excluded from the primary outcome analyses in the per protocol (PP) population.
bRutherford classes 0, 1, and 6 were not part of the protocol eligibility criteria.
cOne patient was enrolled twice, two months apart, with one lesion treated in 1 procedure, and two lesions in the contralateral limb treated in the second procedure. These are included in the count of lesions for the per protocol population (n = 123) and for the ITT population (n = 149).
Continuous data are presented as the means ± standard deviation (range); categorical data are given as the counts (percentage).
Investigational device specifications and procedure
The long flexible catheter shaft of the Phoenix Atherectomy System has a minimum working length of 130 cm. The inward-cutting helical blade sits within a housing that acts as a shield and has an open area to expose the cutter. The cutter is rotated at high speed (10,000 to 12,000 rpm) and shaves material directly into the housing. The debulked material is then conveyed to the proximal part of the catheter by an Archimedes screw on the outside of the torque shaft, which continuously conveys the excised plaque through the handle and into a collection reservoir outside of the patient. The tip of the largest available catheter (2.4 mm) can be deflected to various degrees and rotated so that the cutter can debulk eccentrically to a larger diameter than the catheter itself. The controls for deflection and rotation are housed in the catheter handle with a self-contained battery-powered motor, and the operation of the Phoenix does not require any capital equipment components.
Following the protocol amendment after treatment of the first 36 patients, the use of the 2.2-mm 7F catheter with deflecting tip was discontinued in the trial. The amended protocol also specified that treatment below the level of the popliteal artery was to be limited to use of either 1.8-mm or 2.2-mm catheters (Table 2). For treatment of femoropopliteal lesions, the protocol specified the option of repeating the debulking with a larger catheter if necessary; for target vessel segments below the popliteal artery, the protocol recommended initiating treatment with the 1.8-mm catheter and then employing the 2.2-mm catheter if the RVD was 3.0 to 3.5 mm and additional debulking was required.
Baseline angiographic imaging; device preparation, insertion (standard 0.014-in. guidewire), operation (under fluoroscopic guidance), and removal; and post-procedure PTA or stenting were performed according to the EASE protocol and the Phoenix Instructions for Use. All patients were treated using standard catheterization techniques, with a 6F or larger catheter sheath introducer placed prior to treatment. Anticoagulation consisted of periprocedural aspirin (minimum of 75 mg per day) and clopidogrel (75 mg per day for three days prior or by bolus of 300 or 600 mg); procedural heparin or bivalirudin (to maintain ACT ≥ 250 s, for heparinized patients only); and post-procedural aspirin (minimum of 75 mg per day through six-month follow-up) and clopidogrel (75 mg per day for 30 days or longer at physician discretion). Ticlopidine or prasugrel could be used for patients with known intolerance to clopidogrel.
Follow-up
At 24 h post-procedure, and then at 30 days and six months post-procedure, all patients underwent testing for ABI or TBI, target review of symptoms, a panel of laboratory tests, and assessment for adverse events. The follow-up at 30 days and six months included assessment of Rutherford clinical category and evaluation of clinical success; the 30-day and six-month follow-up also included clinical – and angiographic, if indicated – assessment of need for or outcomes of reintervention including target vessel revascularization (TVR) and target lesion revascularization (TLR). All adverse events were recorded, with notation of severity, duration, action taken, treatment outcome, and relationship to device/procedure, and all serious adverse events (SAEs) were adjudicated by the independent physician reviewer.
Trial endpoints and definitions
The primary efficacy endpoint was technical success, defined as achievement of acute debulking with a post-atherectomy residual diameter stenosis ≤50% (after all Phoenix devices were utilized and prior to any adjunctive therapy), assessed by quantitative angiography or investigator visual estimate. Secondary efficacy endpoints included procedural and clinical success and clinically driven TLR and TVR through six months. Procedural success was defined as the proportion of target lesions in which residual stenosis was ≤30% after treatment with the Phoenix and any other adjunctive therapy. Clinical success was defined as the proportion of patients who had procedural success in all treated lesions with achievement of at least one grade improvement in the Rutherford class evaluation at 30 days and six months.
The primary safety endpoint was freedom from MAEs through 30 days. MAEs were defined as cardiovascular-related deaths; clinically driven TLR; perforations/dissections of grade C or greater that required intervention; symptomatic distal emboli, detected after the index procedure or noted angiographically, and requiring mechanical or pharmacologic intervention; and unplanned amputation. Secondary safety endpoints were the rates of MAEs through six months, adjudicated for seriousness and causality (lesion and device relatedness). Adverse events were considered serious when they resulted in death, were life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, or resulted in persistent or significant disability/incapacity.
Statistical analysis
All statistical summarization and analyses were performed using the PP population (n = 105). The primary safety and efficacy endpoints were analyzed with a one-sided exact binomial test of proportions at a 0.05 overall level of significance to compare the technical success rate and the MAE rate to prespecified target performance goals based on established literature reference rates for other currently marketed atherectomy systems. The primary efficacy objective was to demonstrate that the technical success rate for the Phoenix device is noninferior to that for the comparator atherectomy systems used in similar patient populations. Based on the literature analysis, the endpoint of technical success was to be considered successfully met if the lower limit of the one-sided 95% confidence interval (CI) was above 86%. The primary safety objective was to demonstrate that the proportion of patients experiencing MAEs through 30 days of follow-up is noninferior to that for the comparator systems; the safety endpoint was to be considered successfully met if the upper limit of the one-sided 95% CI was below 20%. No hypothesis testing was performed for the secondary endpoints, for which statistical comparisons used two-sided significance levels of 0.05.
All relevant characteristics were summarized for the PP and ITT analysis populations, with the summary including the mean, standard deviation (SD) with 95% CIs, median, and range for continuous variables, and relative frequencies and 95% CIs for categorical variables. The six-month incidence of clinically driven TVR and TLR was assessed using the Kaplan-Meier method. Statistical analysis was performed using SAS/STAT software, version 9.1 or higher. Planned subgroup analyses included the PP group (n = 105) versus the ITT-only group (n = 23).
Results
The disposition of the trial cohort, including the PP and ITT populations, is diagrammed in Figure 2. At six months, 98 PP patients and 118 ITT patients were evaluable for follow-up, meeting the sample-size requirements determined with the target performance goals for the primary endpoints. Baseline lesion and index procedure details are presented in Table 5 for the PP and ITT populations. In the primary-analysis PP group, 49% (52/105) of patients were treated with the smallest available device (1.8 mm). One patient was pretreated with PTA (Figure 3); in all other procedures, no pretreatment was required.
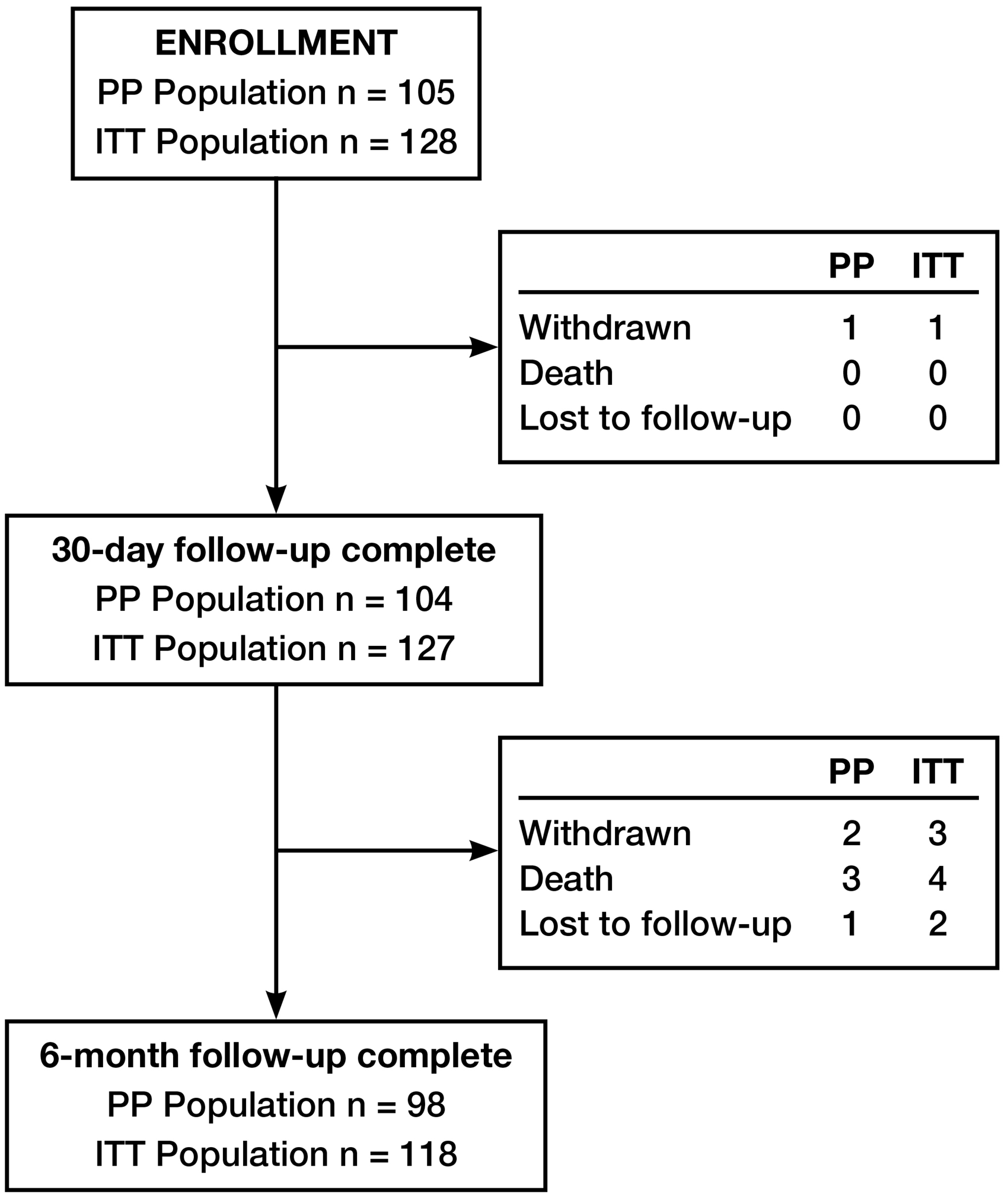
The disposition of the EASE trial cohort, including the per-protocol (PP) and intention-to-treat (ITT) populations.
Baseline lesion and index procedure details.
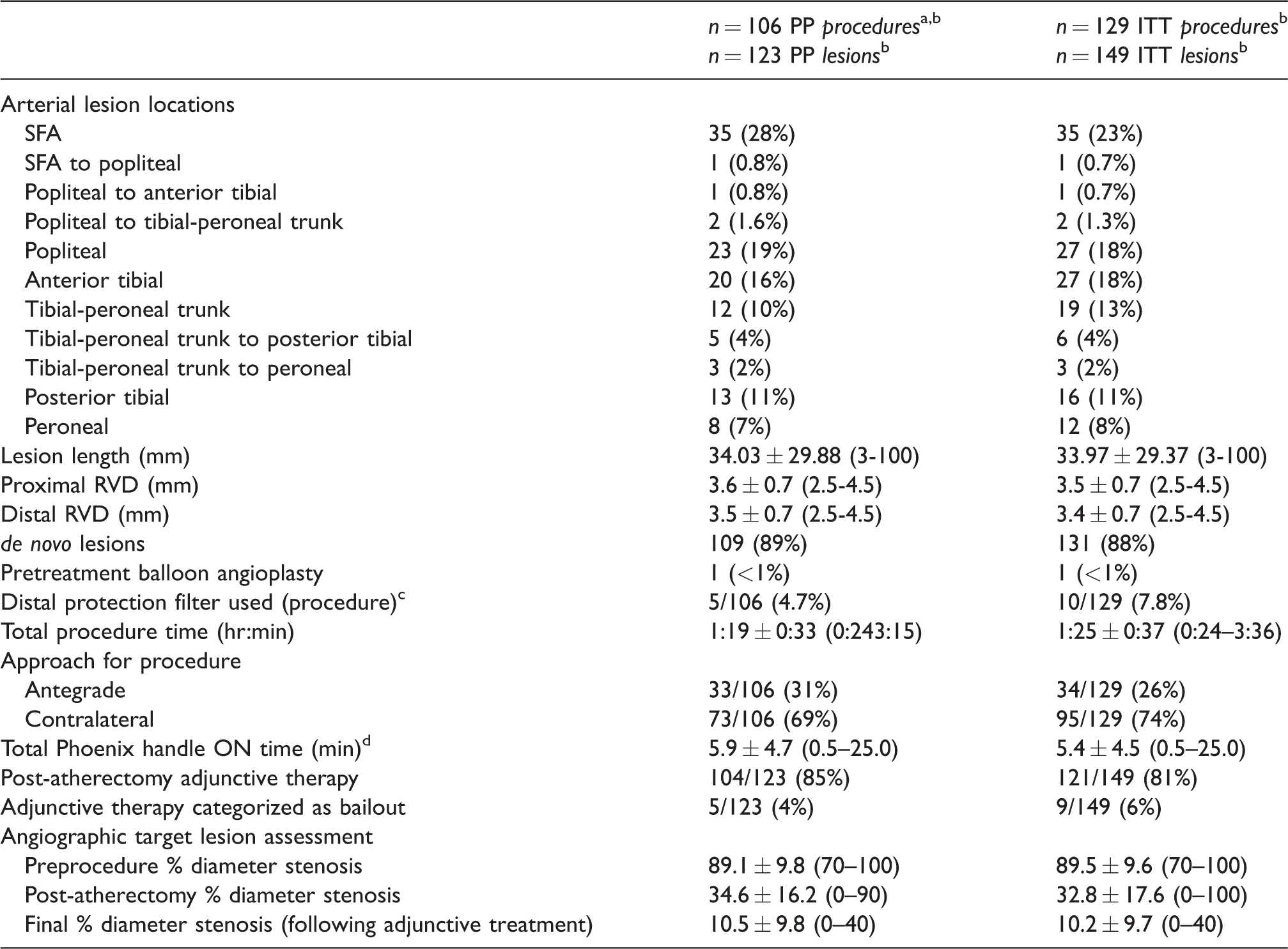
ITT: intention-to-treat; PP: per protocol; RVD: reference vessel diameter; SFA: superficial femoral artery.
aThe original ITT population consisted of 128 patients; but after the first 36 patients had been treated, the protocol guidelines for artery diameter and device catheter sizing relative to anatomical location were amended, and as a result 23 patients were excluded from the primary outcome analyses in the per protocol (PP) population.
bOne patient was enrolled twice, two months apart, with one lesion treated in one procedure, and two lesions in the contralateral limb treated in the second procedure. These are included in the N counts of procedures and lesions for the PP and ITT populations.
cIn all cases, distal protection filters were used during the treatment of non-target lesions. No distal protection filters were used in target lesions treated with the investigational device.
dHandle ON time reflects the total time for the atherectomy portion of the procedure; one or more catheters could be used in treating a target lesion and up to two target lesions could be treated.
Continuous data are presented as the means ± standard deviation (range); categorical data are given as the counts (percentage).
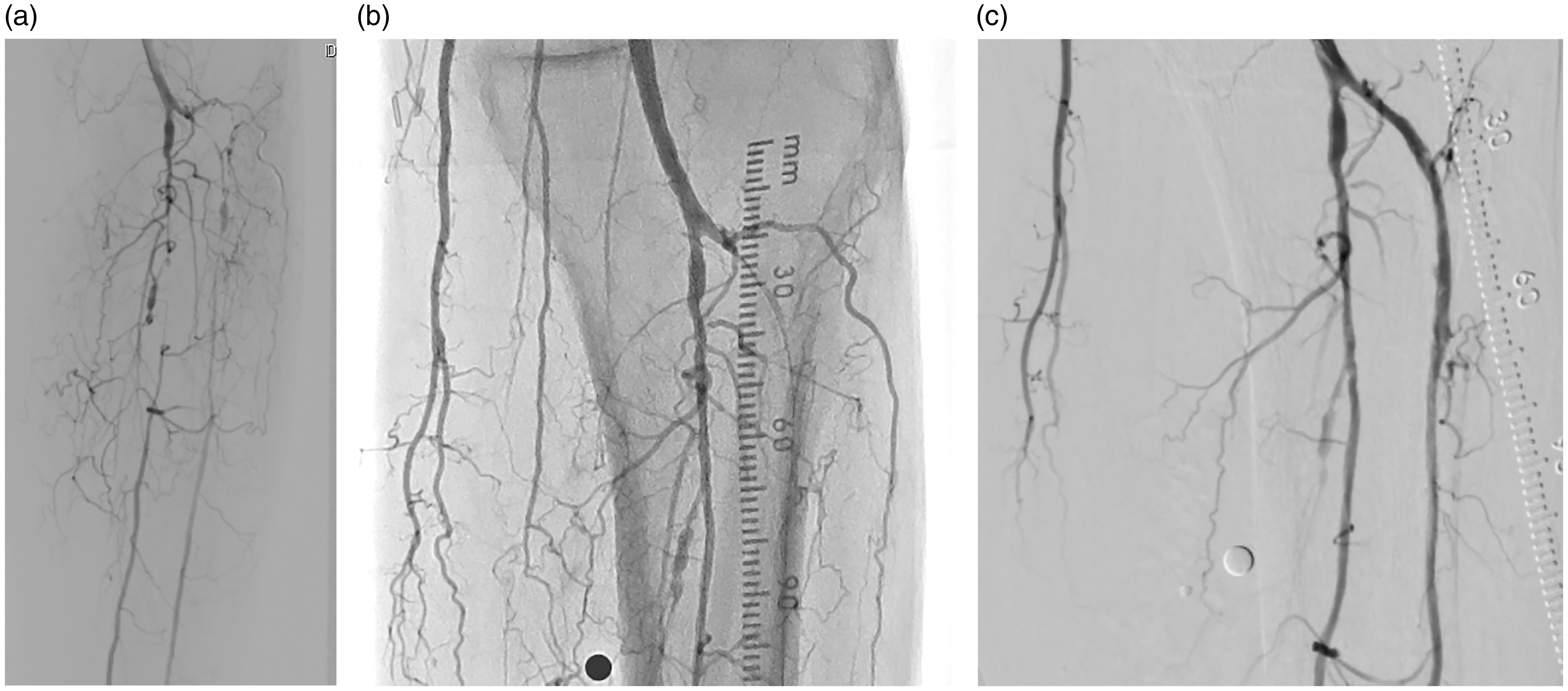
(a) Initial selective below-the-knee (BTK) angiogram showing occlusive disease in peroneal and anterior tibial arteries. (b) The EASE protocol required at least one patent vessel BTK, so balloon angioplasty was performed initially on the peroneal artery to restore flow. (c) Stand-alone atherectomy was subsequently performed in the anterior tibial artery with the Phoenix Atherectomy System.
Post-atherectomy adjunctive therapy was used to complete treatment of 81% (121/149) of the target lesions in the ITT group and 85% (104/123) of the target lesions in the PP group. The post-atherectomy adjunctive therapy used to complete treatment in the PP group included PTA in 91% (95/104) of these cases, cutting balloon angioplasty in 12% (12/104), stenting in 1% (1/104), and atherectomy in 2% (2/104). In five of those cases in the PP group, the adjunctive therapy was categorized as “bailout,” although in four of the five cases, the residual stenosis following the initial atherectomy portion of the procedure was <50%, thereby qualifying the cases as technical successes. The reasons for bailout were dissection in three of those cases (one of which was flow-limiting, all treated with PTA) and perforation in two (1 treated with PTA, 1 treated with PTA and a stent). In all cases that required bailout (the five cases in the PP group and another four in the ITT-only population), a final diameter stenosis of ≤30% met the definition of procedural success. The mean final diameter stenosis was 10.5% ± 9.8% in the primary analysis PP group and 10.2% ± 9.7% in the ITT group.
Efficacy outcomes
The outcomes for the primary and secondary efficacy endpoints are summarized in Table 6 for the PP and ITT populations. The primary technical success endpoint was achieved for 95.1% (117/123) of lesions in the primary analysis PP group, with ≤50% residual stenosis after acute debulking. The lower limit of the 95% CI was 90.6%, so the target performance goal (≥86%) was successfully met. The target performance goal was also met for the ITT group, in which there was one additional case (beyond those tabulated for the PP group) in which technical success was not achieved. In all seven cases in which technical success was not achieved, the secondary endpoint of procedural success (≤30% residual stenosis) was achieved with adjunctive intervention performed. The rate of procedural lesion success was 99.2% (122/123) for the primary analysis PP group and 98.7% (147/149) for the ITT group. In the single procedural failure in the PP group, the target lesion had a residual stenosis of 50% after debulking with the investigational device, but after adjunctive PTA, the residual stenosis was reduced to only 40%. For this patient, no procedural complications or adverse events were observed through the six-month follow-up.
Primary and secondary efficacy endpoint outcomes.
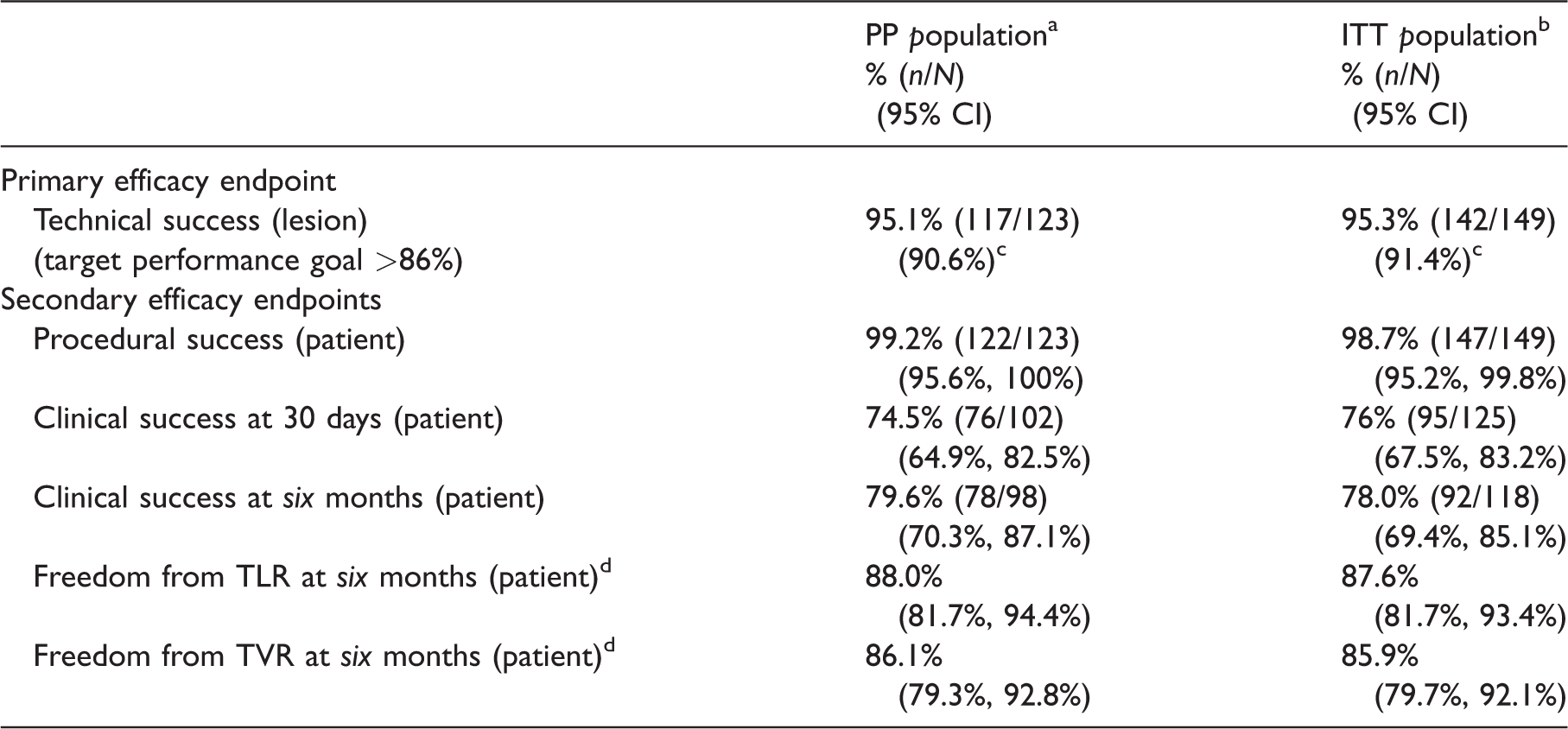
CI: confidence interval; TLR: target lesion revascularization; TVR: target vessel revascularization.
Technical success: The achievement of acute debulking with a post-atherectomy residual diameter stenosis ≤50% (after all Phoenix devices were utilized and prior to any adjunctive therapy), assessed by quantitative angiography or investigator visual estimate.
Procedural success: The proportion of target lesions in which residual stenosis was ≤30% after treatment with the Phoenix and any other adjunctive therapy.
Clinical success: The proportion of patients who had procedural success in all treated lesions with achievement of at least one grade improvement in the Rutherford class evaluation at 30 days and six months.
aPer protocol (PP) population: N = 105 patients/123 lesions.
bIntention-to-treat (ITT) population: N = 128 patients/149 lesions.
cFor the primary efficacy endpoint: exact binomial lower one-sided 95% CI.
d Kaplan-Meier estimates.
Clinical success was achieved at 30 days for 74.5% of the PP group and 76% of the ITT group, and at six months for 79.6% of the PP group and 78% of the ITT group, reflecting the sustained improvement in Rutherford classification. All patients had either the same or better Rutherford class symptoms at 30 days, and only three patients had a decrease in Rutherford class at the six-month follow-up (Table 7). For the PP group, the mean ABI increased from 0.70 ± 0.23 at baseline to 0.96 ± 0.21 (n = 98) at 30 days and 0.84 ± 0.29 (n = 88) at six months. For the ITT group, the mean ABI increased from 0.70 ± 0.22 at baseline to 0.95 ± 0.20 (n = 119) at 30 days and 0.82 ± 0.28 (n = 107) at six months. For the PP group, the Kaplan-Meier estimates of freedom from TLR and freedom from TVR at six months were 88.0% (95% CI: 81.7%–94.4%) and 86.1% (95% CI: 79.3%–92.8%), respectively. For the ITT group, the Kaplan-Meier estimates of freedom from TLR and freedom from TVR at six months were 87.6% (95% CI: 81.7%–93.4%) and 85.9% (95% CI: 79.7%–92.1%), respectively.
Rutherford class changes through six months.
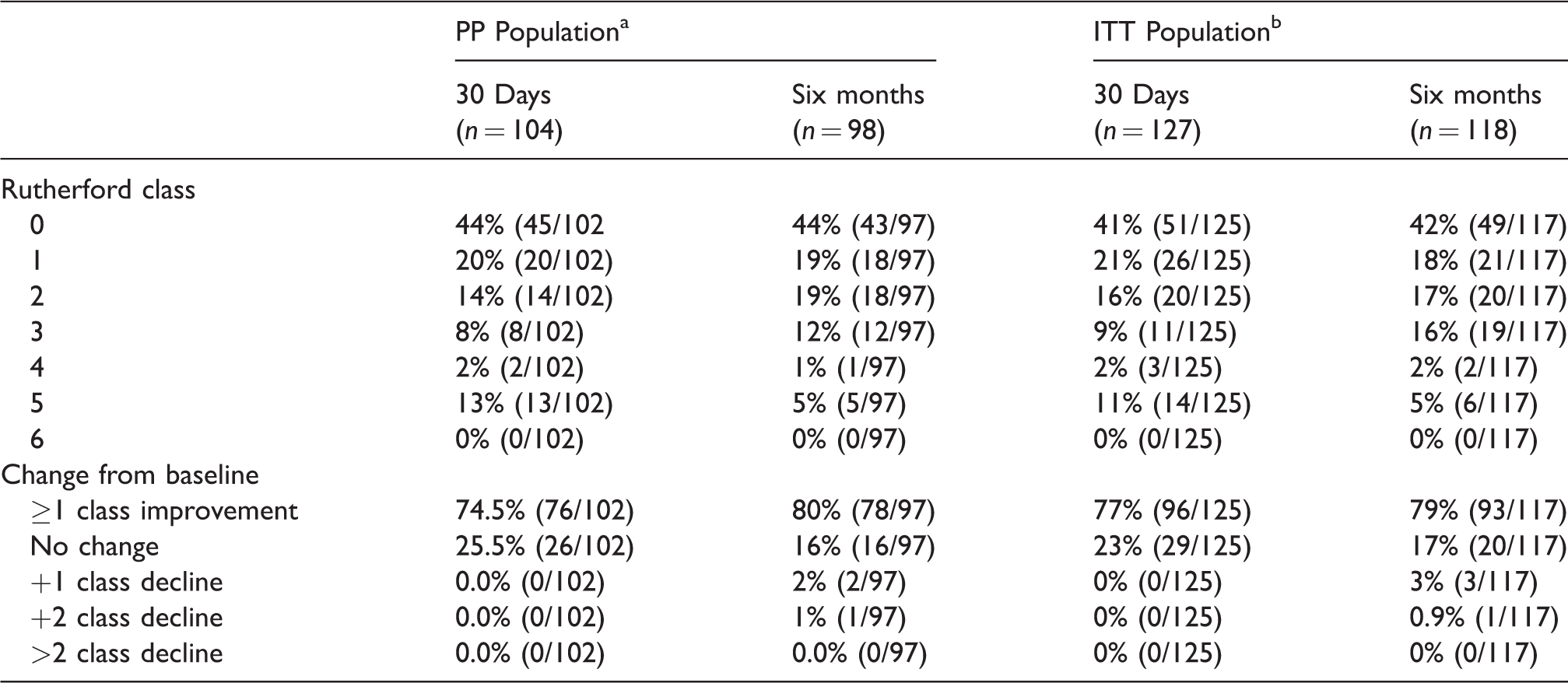
aPer protocol (PP) population: N = 105 patients/123 lesions.
bIntention-to-treat (ITT) population: N = 128 patients/149 lesions.
Safety outcomes
The outcomes for the primary and secondary safety endpoints are summarized in Table 8 for the PP and ITT populations. Through the 30-day follow-up, MAEs were experienced by 5.7% (6/105) of patients in the primary analysis PP group and by 9.4% (12/128) of patients in the ITT group. With an upper one-sided CI of 11.0%, the target performance goal (<20%) was successfully met for the PP group. The target performance goal was also met for the ITT group, with an upper one-sided CI of 14.8%.
Primary and secondary safety endpoint outcomes.
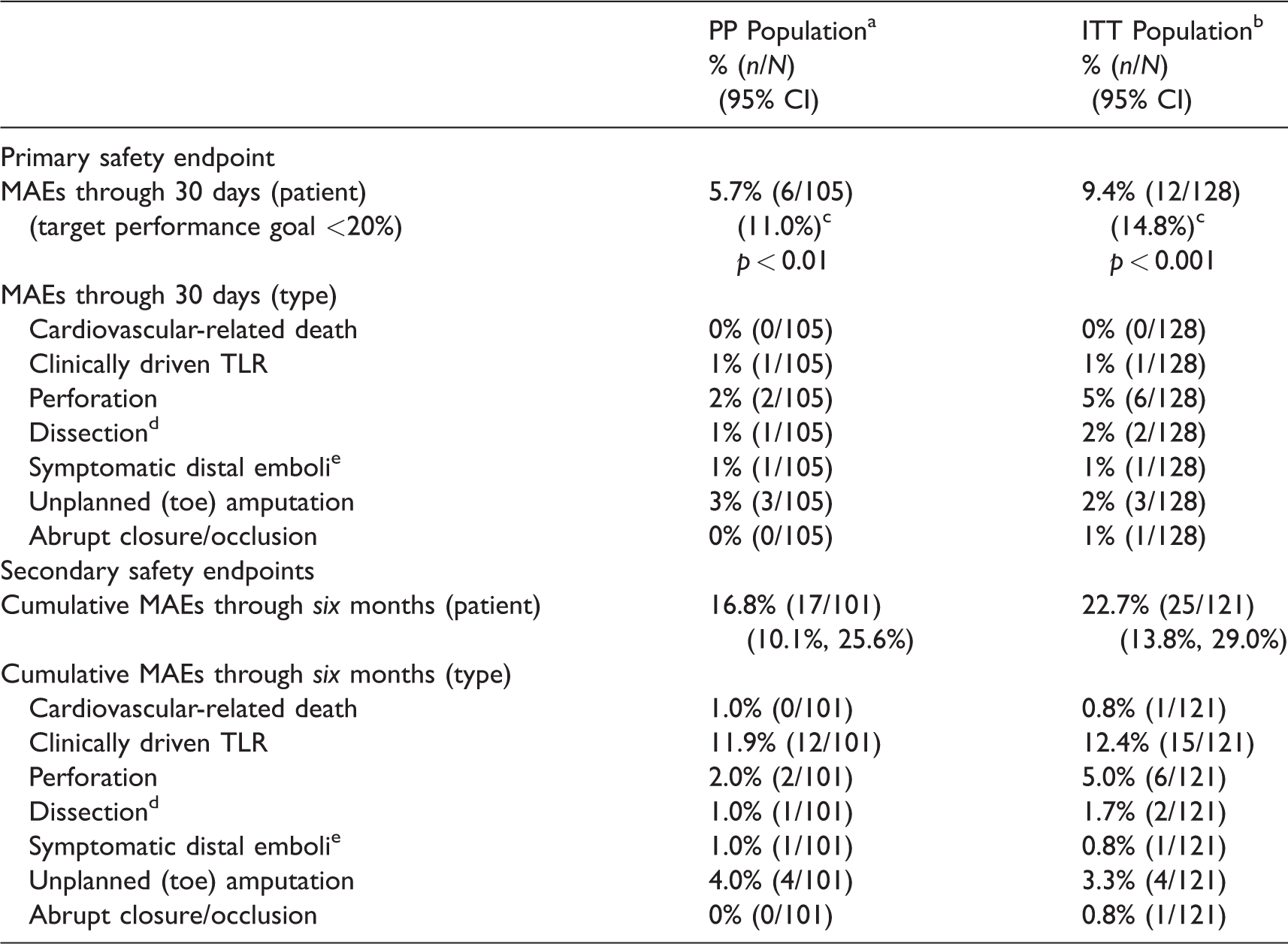
CI: confidence interval; MAE: major adverse event; TLR: target lesion revascularization.
aPer protocol (PP) population: N = 105 patients/123 lesions.
bIntention-to-treat (ITT) population: N = 128 patients/149 lesions.
cFor the primary safety endpoint: exact binomial upper one-sided 95% CI.
dGrade C or greater that require an intervention to resolve.
eClinical signs or symptoms of distal emboli detected in treated limb distal to the treated lesion after the index procedure and requiring mechanical or pharmacological means to improve flow.
Through 30 days, there were eight MAEs in six PP patients, and 14 MAEs in 12 ITT patients. A single PP patient had three MAEs – procedurally, a flow-limiting dissection and an embolus, both resolved with PTA; and an unplanned toe amputation, secondary to osteomyelitis, prior to the 30-day follow-up. This was the only case of distal embolization in the full trial cohort.
Through six-month follow-up, MAEs were experienced by 16.8% (17/101) of PP patients (21 MAEs altogether) and by 22.7% (25/121) of ITT patients (30 MAEs altogether). The cumulative increase between the 30-day and six-month follow-up visits was primarily driven by the need for reintervention in the target lesion – with the rate of TLR increasing from 1% (1/105) at 30 days to 11.9% (12/101) at six months in the PP group, and from 1% (1/128) at 30 days to 12.4% (15/121) at six months in the ITT group. Through six months, there were four deaths, all occurring in the interval after the 30-day follow-up. One death at 153 days post-procedure was the result of a myocardial infarction and was determined to be cardiovascular-related and hence an MAE, but not related to the investigational device or procedure. The other three deaths – occurring at 39 days due to septic shock, at 81 days due to lymphoma, and at 144 days due to chronic obstructive pulmonary disease – were not considered MAEs.
Through six months, a total of 125 adverse events occurred in 62 patients in the PP population, with 28 events related to the target lesion and 20 of those events determined to be also related to the investigational device – including arterial restenosis in seven cases, type A or B dissection in four, intermittent claudication in three, vessel perforation in two, and one case each of type C dissection, in-stent arterial restenosis, stent occlusion, and skin ulcer. A total of 195 adverse events occurred in 81 patients in the ITT population, with 43 events related to the target lesion and 32 of those events determined to be also related to the investigational device. Overall in the ITT population, a total of 129 of the adverse events were classified as serious, and 26 of those were related to the target lesion while 23 were also related to the investigational device. In the PP population, a total of 83 of the adverse events were classified as serious, and 14 of those were related to the target lesion and to the investigational device. Non-flow-limiting dissections (type A or B) were the most frequent target-lesion-related events (11/28 in the PP group and 14/43 in the ITT group, with none being MAEs). Other target-lesion-related events included arterial restenosis (7/28 in the PP group with six being MAEs, and 10/43 in the ITT group with nine being MAEs), intermittent claudication (4/28 in the PP group with three being MAEs, and 4/43 in the ITT group with three being MAEs), and vessel perforation (2/28 in the PP group with both being MAEs, and 7/43 in the ITT group with six being MAEs).
Subgroup analyses
The primary and secondary endpoints for efficacy and safety were compared between the primary analysis PP population and the ITT-only population of 23 patients who did not meet the amended trial eligibility criteria, in that they were treated according to the discontinued vessel/device sizing regimen. While the proportion of patients with MAEs was significantly lower for the PP group than for the ITT-only group at both 30 days (p = 0.008) and six months (p = 0.032), no other endpoints showed a significant difference. Freedom from TLR (p = 0.668) and TVR (p = 0.877) was similar between the PP and ITT-only groups, suggesting that the impact of the early MAEs associated with the discontinued regimen did not persist in terms of reintervention requirements.
In the subgroup analysis comparing CLI patients versus non-CLI patients, the achievement of clinical success was significantly greater for the non-CLI patients in both the PP group (59% vs. 81%, p = 0.018) and the entire ITT group (64% vs. 82%, p = 0.029) at 30-day follow-up. Reflecting across-the-board improvements in Rutherford class, the difference in achievement of clinical success was no longer significant at the six-month follow-up, with all rates above 74%. In a subgroup analysis of US vs. German sites, the rate of clinical success was also significantly different, favoring the US sites, for both the PP (p = 0.043) and ITT (p = 0.026) populations at the 30-day follow-up, but the significant difference did not persist to the six-month follow-up.
When the proportions of patients experiencing MAEs at 30 days were compared across sites using Fisher’s exact test, no significant difference was detected for either the PP group (p = 0.94) or the ITT group (p = 0.86). When the primary safety endpoint was compared between the US sites as one group and the German sites as another group, again no significant differences were detected. The appropriateness of the across-site pooling of results for analysis is thus confirmed.
Discussion
With independent medical-reviewer adjudication, the EASE trial evaluated the performance of the novel single-insertion Phoenix Atherectomy System in first-line treatment of infrainguinal PAD in a diverse and challenging population of patients (32% with CLI, 53% with diabetes mellitus) and presentation of lesions (mean diameter stenosis 89%, approximately 70% at or below the knee).
Atherectomy devices for peripheral vascular indications are cleared for commercial distribution in the United States by a 510(k) process, which has been designed by the FDA to establish substantial equivalence to predicate devices already cleared for use. The 510(k) process includes a comprehensive submission with data covering bench, preclinical, and clinical work. The clinical data consist of a single-arm study based on approved prespecified endpoint performance goals using data from previous clinical studies with similar devices. All of the data – bench, preclinical, and clinical – are used to establish the safety and effectiveness of a device relative to predicate devices. The Phoenix Atherectomy System has undergone the 510(k) process, which included the single-arm EASE IDE trial, and received FDA clearance in January 2014.
In terms of the device’s efficacy in decreasing arterial stenosis, the primary technical success endpoint (≤50% residual stenosis prior to adjunctive therapy) was achieved by 95.1% of patients in the PP group, and the lower limit of the 95% CI was 90.6%, successfully meeting the target performance goal of ≥86%. Following post-atherectomy adjunctive treatment – which consisted of PTA in 91% of cases and bailout stenting in only 1 case – post-procedure residual diameter stenosis ≤30% was achieved for all but one of the PP patients, and mean final diameter stenosis was 10.5%. The protocol for the EASE trial allowed for adjunctive therapy to be used after the atherectomy portion of the procedure, as it is not uncommon to limit debulking of a lesion and then complete the procedure with an adjunctive therapy. In terms of clinical success, the goal of ≥1 Rutherford class improvement was achieved for 75% of patients at 30 days and 80% at six months, confirming the durability of the clinical results, and six-month freedom from clinically driven TLR and TVR was 88% and 86%, respectively.
In terms of device safety, the rates of adverse events and their frequencies were within the expected ranges, and there were no unanticipated adverse events during the course of the study. Through 30 days, the proportion of patients in the primary analysis PP group experiencing MAEs was 5.7%, and the upper limit of the one-sided 95% CI was 11.0%, meeting the target performance goal of <20%. Through six months, a total of 21 MAEs were experienced by 16.8% (17/101) of patients in the PP group. One death, occurring at 153 days post-procedure, the result of a myocardial infarction, was determined to be cardiovascular-related and hence an MAE, although not related to the investigational device or procedure. The cumulative increase in MAEs between 30 days and six months was primarily the result of the need for TLR, which was required for 11 patients during that interval.
Plaque excision techniques have been employed in the treatment of PAD for at least 25 years. 24 While balloons and stents function by pushing plaque into the vessel wall, catheter-deliverable atherectomy devices debulk and remove atherosclerotic plaque by cutting, pulverizing, or shaving.10,18 Atherectomy has been used as part of an overall treatment strategy for endovascular revascularization – either in a stand-alone capacity or as a means of preparing the arterial lumen for PTA and/or stenting.18,19 Over time, various devices with different mechanisms of action have been developed and approved for use in the peripheral vasculature, including directional atherectomy (plaque excision), laser ablation, rotational atherectomy (shaving and aspirating), and orbital atherectomy (pulverizing). The approved atherectomy devices have shown good technical success in procedural reduction of stenosis, effectiveness in reducing PAD symptoms, and acceptable but varying levels of adverse events.13,14,17 All modalities of peripheral endovascular intervention can lead to mechanical injury – beginning with the disruption or removal of the endothelial layer during catheterization and balloon dilation – inducing vascular inflammation, which stimulates smooth muscle cell proliferation and extracellular matrix deposition, with neointimal thickening and restenosis.18,25,26 With options including PTA, self-expanding nitinol stents, and drug-eluting stents and balloons as well as the atherectomy modalities, the choice of an optimal endovascular strategy varies in given situations in relation to specific features of the peripheral vascular bed and the diffuse nature of the atherosclerotic process. 18
The design of the Phoenix Atherectomy System is distinct from that of the currently approved directional, rotational, and orbital atherectomy systems in ways that may affect ease of use and the efficacy and safety of device performance. The Phoenix debulks plaque with a front-cutting blade, then captures the plaque within the device shaft, and continuously clears the debris by means of the mechanical operation of an Archimedes screw. This mechanism offers the ability to debulk arteries that previously may not have been candidates for atherectomy. The incorporation of a directable deflecting tip in the largest Phoenix catheter allows it to create a lumen larger than the catheter diameter, whereas the 1.8-mm and 2.2-mm catheters are suited for vessels down to the ankle. The front (distal) positioning of the blade means that there is no need to pass a nosecone through the disease segment prior to treatment, as is the case with the nose cone of the SilverHawk and TurboHawk devices (Covidien, Plymouth, MN). Unlike these directional devices, which must be withdrawn periodically for removal of debris from the nose cone, since the Phoenix continually captures and clears debulked material, it can perform an entire debulking procedure with one single insertion; or sequential single insertions can be employed with catheters of progressively increasing diameter. Unlike the Jetstream rotational device (Pathway Medical, Kirkland, WA), the strictly mechanical clearance featured by the Phoenix avoids the potential consequences of aspiration, including excessive blood removal and vessel suck-down. The operation of the Phoenix requires no capital equipment component or tableside infusion pump, as with the Spectranetics laser, the Jetstream device, or the Orbital Atherectomy System (Cardiovascular Systems, St. Paul, MN).
The performance of the Phoenix Atherectomy System in the EASE trial compares favorably with the performance of the other three currently available atherectomy modalities, as reported in recent clinical trials. The procedural success (≤30% residual stenosis) of 99.2% matched that in the multicenter single-arm Pathway PVD trial (rotational atherectomy with aspiration), 14 with a higher use of adjunctive PTA (90% vs. 59%) but a lower use of adjunctive stenting (<1% vs. 7%). The procedural success rate in the multicenter single-arm DEFINITIVE-LE trial (SilverHawk and TurboHawk directional atherectomy) was 87%, with 35% of patients receiving adjunctive post-atherectomy therapy, including stenting in 3.2%. 13 The rates of periprocedural flow-limiting dissection and perforation, respectively, were 1% and 2% in the EASE trial, 9% and 2% in the Pathway PVD trial, and 2.3% and 5.3% in the DEFINITIVE-LE trial. Whereas the mean final post-procedure (post-adjunctive) diameter stenosis was 10.5% in the EASE trial, it was 18.6% in the DEFINITIVE-LE trial, 13 21.4% in the Pathway PVD trial, 14 and 10% to 11% in the single-arm CONFIRM registry series (Orbital Atherectomy System). 17 The six-month rate of TLR was 11.9% in the EASE trial and 15% in the Pathway PVD trial. 14 At six months, 80% of patients in the EASE trial experienced an improvement of ≥1 Rutherford class, compared to 68% of patients in the Pathway PVD trial. 14
The expressed intention of preventing distal embolization with the Phoenix mechanism of action was substantially realized in the EASE trial. Distal protection was used in 4.7% of procedures in the PP group, but none of these embolic protection devices were used in the treatment of target lesions. Even with no use of distal protection, there was only one instance (1.0%) of distal embolization during the trial, occurring procedurally and resolved with PTA. The embolization rate reported for the DEFINITIVE-LE trial of the SilverHawk and TurboHawk devices was 3.8%, 13 the rate of “minor embolizations” in the Pathway PVD trial of the Jetstream was 10%, 14 and the rate reported in the prospective multicenter CONFIRM registry for the Orbital Atherectomy System was 2.2%. 17
EASE was a single-arm regulatory IDE trial conducted to establish substantial equivalence to already cleared predicate devices, based on comparison with prespecified performance goals on approved endpoints. As such, like the comparator device studies, the trial was not randomized. Since follow-up imaging was only performed as needed for decision making about and outcome evaluation of clinically driven revascularization, the rates of binary restenosis and primary/secondary patency were not calculated. While no subgroup analysis was included on baseline diabetes status, as in the DEFINITIVE-LE trial of directional atherectomy, 13 the EASE trial included a similar number of patients with diabetes at baseline (approximately 50%) and, as noted, achieved comparable overall results. Because only the largest of the available Phoenix catheter sizes had the capability of deflection, the absolute volume of plaque removed was limited, suggesting that at present this initial generation of the device may be better suited for the popliteal artery or below. Because of the trial’s limitation in terms of debulking severe calcification, clear evidence was not provided concerning the extent of calcium ablation achievable with the device. At the same time, as the device is designed to capture more rigid plaque elements, highly mobile content such as fresh thrombus has not yet been studied and may still pose embolic risks to the distal vasculature.
Conclusion
The EASE trial confirmed the safety and effectiveness of first-line treatment with the Phoenix Atherectomy System through six months in a range of ATK and BTK arterial locations and diameters, PAD categories, and disease morphologies. With ≤50% residual stenosis achieved for 95.1% of patients after acute debulking and prior to adjunctive therapy, and with only 5.7% of patients experiencing MAEs through 30 days, the target performance goals for the primary endpoints were met, supporting the substantial equivalence of the Phoenix with other available devices for atherectomy treatment of infrainguinal PAD. Additional real-world and comparative studies with longer follow-up are needed, including evaluation of the device utility in preparing the vessel for drug-delivery modalities.
Footnotes
Acknowledgments
The authors acknowledge the contribution of James F. McKinsey, MD, of Columbia Presbyterian Hospital in New York City, as co-principal investigator of the trial along with the lead author. We also thank the physicians who participated as study site investigators. Independent study monitors were provided by MedPass; independent data management services were provided by Innoventz Corporation; biostatistical support was provided by Boston-Biomedical Associates and by Tami Crabtree, MS. Boston-Biomedical Associates provided independent medical review and physician adjudication of major adverse events (MAEs), serious adverse events (SAEs), device-related events, and procedure-related events.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Davis and Ramaiah have each done consulting work for Volcano Corporation. The other authors have no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The EASE study was funded by Volcano Corporation.