
Abstract
In many nanotechnology areas, there is often a lack of well-formed conceptual ideas and sophisticated mathematical modeling in the analysis of fundamental issues involved in atomic and molecular interactions of nanostructures. Mathematical modeling can generate important insights into complex processes and reveal optimal parameters or situations that might be difficult or even impossible to discern through either extensive computation or experimentation. We review the use of applied mathematical modeling in order to determine the atomic and molecular interaction energies between nanoscale objects. In particular, we examine the use of the 6-12 Lennard-Jones potential and the continuous approximation, which assumes that discrete atomic interactions can be replaced by average surface or volume atomic densities distributed on or throughout a volume. The considerable benefit of using the Lennard-Jones potential and the continuous approximation is that the interaction energies can often be evaluated analytically, which means that extensive numerical landscapes can be determined virtually instantaneously. Formulae are presented for idealized molecular building blocks, and then, various applications of the formulae are considered, including gigahertz oscillators, hydrogen storage in metal-organic frameworks, water purification, and targeted drug delivery. The modeling approach reviewed here can be applied to a variety of interacting atomic structures and leads to analytical formulae suitable for numerical evaluation.
Keywords
Introduction
For the past two decades, nanotechnology has been a major focus in science and technology. However, in various areas of physics, chemistry, and biology, both past and current research involving interacting atomic structures are predominantly either experimental or computational in nature. Both experimental work and large-scale computation, perhaps using molecular dynamics simulations, can often be expensive and time-consuming. On the other hand, applied mathematical modeling often produces analytical formulae giving rise to virtually instantaneous numerical data. This can significantly reduce the time taken in the trial-and-error processes leading to applications and which in turn significantly decreases the research cost. Here, applied mathematical modeling in nanotechnology is reviewed, and particularly, the work of the present authors and their colleagues in the use of classical mathematical modeling procedures to investigate the mechanics of interacting nanoscale systems for various applications, including nano-oscillators, metal-organic frameworks (MOFs), molecular selective separation, and drug delivery.
Throughout, the dominant mechanisms behind these nanoscale systems are assumed to arise from atomic and molecular interactions that can be modeled by the 6-12 Lennard-Jones potential function (see equation (4)), and further simplifications are made by adopting the continuous or continuum assumption. This approximation assumes that two interacting molecules can be replaced by two surfaces or two regions, for which the discrete atomic structure is averaged over the surface or the volume with a constant atomic surface density or a constant atomic volume density, respectively. Basically, the continuous assumption gives an average result, and it is much better suited to those situations involving well-defined surfaces with evenly distributed atoms, such as graphene, carbon nanotubes, or carbon fullerenes. In each of these instances, there exists a uniform distribution of atoms, and the continuous approximation might be most accurate. In the case of non-evenly distributed atomic structures, a hybrid approach is adopted, which deals with the isolated atoms individually, and the continuous approximation is adopted for the remainder. For example, a methane molecule CH4 is assumed to be replaced by a spherical surface of a certain radius with a constant hydrogen atomic surface density, together with a single carbon atom located at the center of the spherical surface.1,2
In this review, we comment that we do not include the mechanics of dislocations in metallic materials or the use of the Cauchy–Born rule to bridge interactions since the modeling here assumes that there is no deformation of any surface due to the van der Waals interactions. We refer the reader to Van der Giessent and Needleman 3 for a comprehensive study of plastic discrete dislocations and to Biner and Morris 4 for a computational simulation of the discrete dislocation method. Furthermore, a review of the Cauchy–Born rule can be found in Ericksen. 5
In the following section, both the 6-12 Lennard-Jones potential function and the continuous approximation are introduced. In the section thereafter, analytical expressions are presented for the interaction energies of the basic molecular building blocks, namely, points, lines, planes, rings, spheres, and cylinders, all deduced utilizing the 6-12 Lennard-Jones potential function together with the continuous approximation. In the section on the mechanics of nanostructures, the mechanics of the so-called gigahertz oscillators is reviewed, including the determination of the energy and force distributions of this nanostructured device. The development of a mathematical model of MOFs for gas storage is presented in the section thereafter. In the next section, the modeling approach is reviewed for molecular selectivity and separation for water purification, ion separation, and biomolecule selection. In the targeted drug delivery section, we present a review of applied mathematical modeling for targeted drug delivery. A brief overall summary is presented in the final section of this article.
Lennard-Jones atomic interaction potential and the continuous approach
For two separate non-bonded molecular structures, the interaction energy E can be evaluated either directly using a discrete atom–atom formulation or approximately using the continuous approach. Thus, the non-bonded interaction energy may be obtained either as a summation of the individual interaction energies between each atomic pair, namely

where

where
The hybrid discrete–continuous approach applies to the modeling of irregularly shaped molecules, such as drugs, and constitutes an alternative approximation to determine the interaction energy. The hybrid approach is represented by elements of both equations (1) and (2) and can be effective when a symmetrical molecule is interacting with a molecule comprising asymmetrically located atoms. In this case, the interaction energy is given as follows

where
The continuous approach is an important approximation, and Girifalco et al.
6
state that
From a physical point of view the discrete atom-atom model is not necessarily preferable to the continuum model. The discrete model assumes that each atom is the center of a spherically symmetric electron distribution while the continuum model assumes that the electron distribution is uniform over the surface. Both of these assumptions are incorrect and a case can even be made that the continuum model is closer to reality than a set of discrete Lennard-Jones centers.
One such example is a
The continuum or continuous approximation has been successfully applied to a number of systems, including the interaction energy between nanostructures of various types and shapes, namely, carbon fullerenes,6,10,11 carbon nanotubes,6,12–21 carbon nanotube bundles,22–24 carbon nanotori,25–30 carbon nanocones,31–34 carbon nanostacked cups, 35 fullerene–nanotube,8,36–46 and TiO2 nanotubes.47–49 Moreover, this method has also been used in systems involving proteins and enzymes,50–52 DNA,52–55 lipid bilayer and lipid nanotube,56–58 water molecule,59–64 benzene,2,65–69 methane,2,3,70–75 ions,75–79 and gas storage and porous aromatic frameworks.80–88
The Lennard-Jones potential function

where
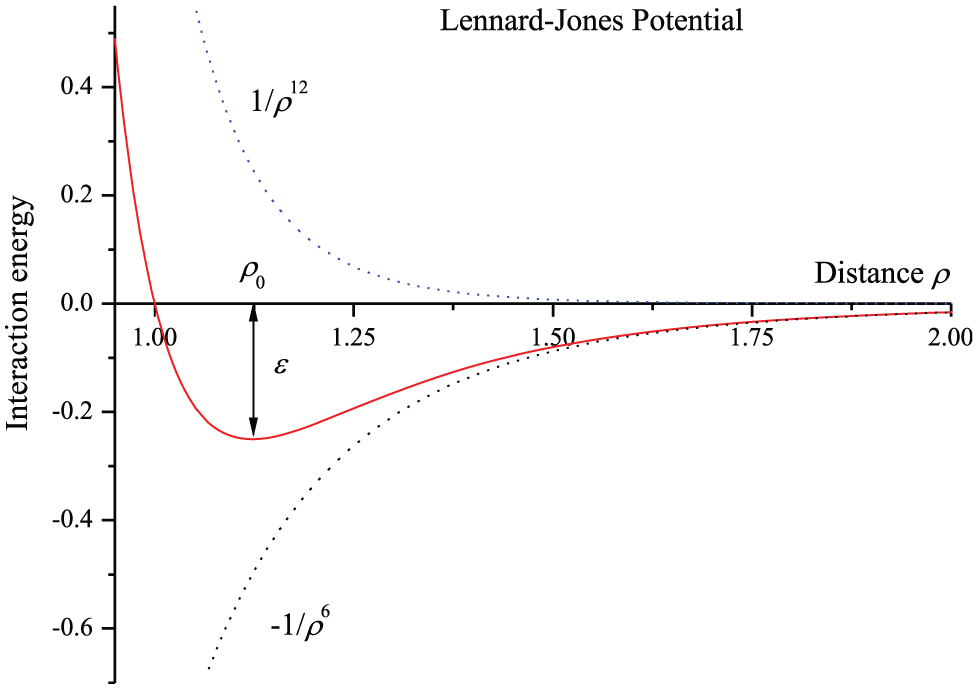
Lennard-Jones potential.
Numerical values for the Lennard-Jones constants taken from Mayo et al. 90

When the Lennard-Jones potential function

where

must be evaluated. In many instances, integrals of this type can be given explicitly in terms of the hypergeometric function

provided that
Analytical expressions for idealized molecular building blocks
In this section, the approach adopted by Thornton and colleaugues80–82 and Lim et al. 87 is summarized using idealized building blocks to represent the interactions of both simple and more complicated geometries of nanostructures yielding simple and elegant analytical models. First, the analytical representations of the van der Waals interaction between an atom and the building blocks, which are represented by standard geometrical shapes such as points, lines, planes, rings, spheres, and cylinders are determined. At first sight, such a dramatically simplified modeling approach may seem geometrically severe, but in many situations, it has been shown to provide the major contribution to the interaction energy of the actual structure.
Interaction of two atomic points
Given the coordinates of two atoms,
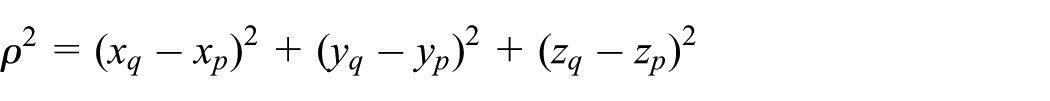
Interaction of atomic point with atomic line
The perpendicular (closest) distance between an atomic point and an atomic line is denoted by


Interaction of atoms with idealized building blocks: (a) point with line, (b) point with plane, (c) point with ring, (d) point with spherical surface, and (e) point with infinitely long right-cylindrical surface.
On making a change of variable and substituting

which can then be evaluated using

to obtain

Interaction of atomic point with atomic plane
This situation is relevant to modeling nanostructures as it corresponds to the case of an individual atom interacting with a graphene sheet. Again, the perpendicular spacing between the point and the plane is assumed to be

The substitution
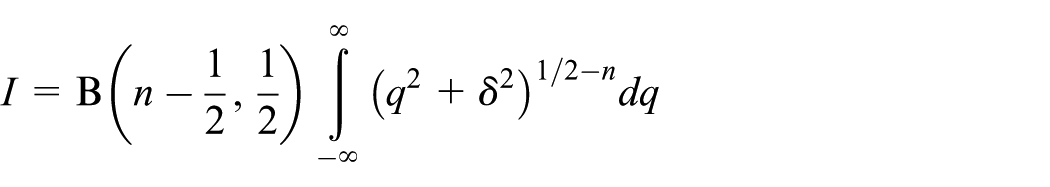
On making a further substitution of
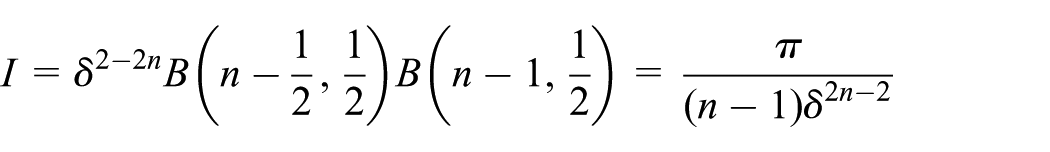
Interaction of atomic point with atomic ring
The interaction of a point with a ring can be categorized into two cases which are as follows: (1) the point is interacting with the ring from the side and (2) the point is interacting with the ring from the top or bottom. For the first case, the point

On making the substitution
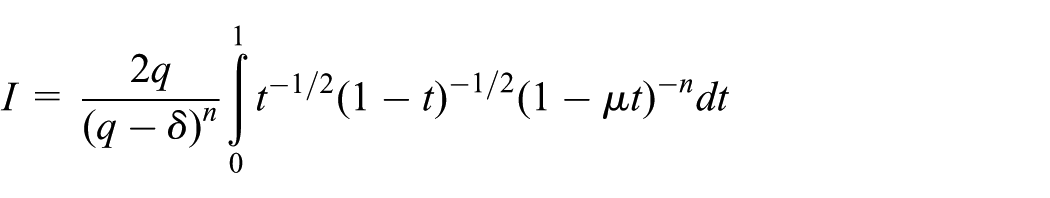
where

The Pfaff transformation is utilized,
94

In the case of an atomic point
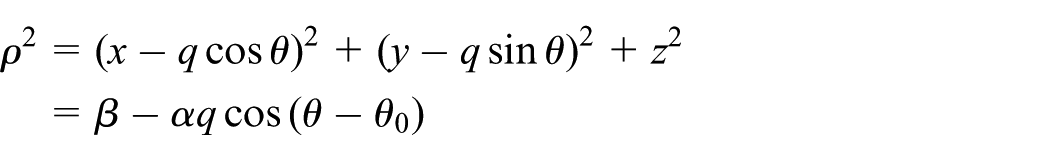
where
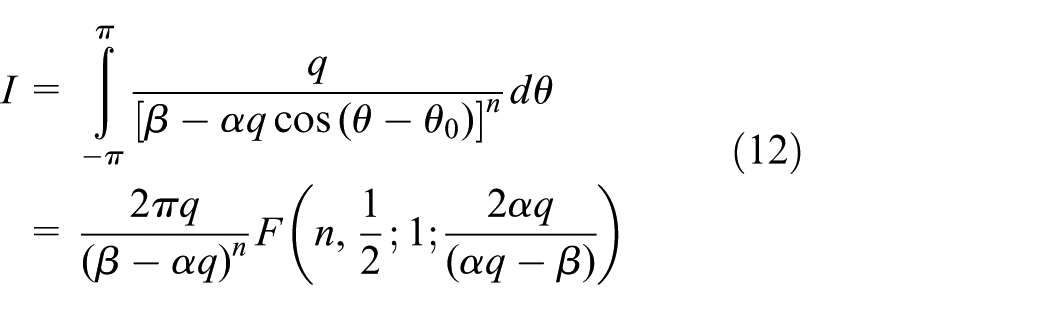
Interaction of atomic point with atomic spherical surface
The atomic point with Cartesian coordinates
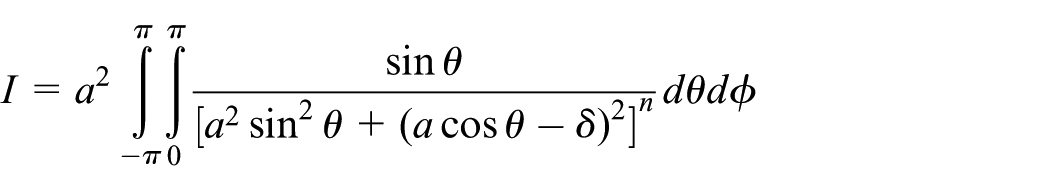
Since the integrand in this case is independent of

which on making the substitution

Interaction of atomic point with infinite atomic cylindrical surface
Here, the interaction of an arbitrary atomic point
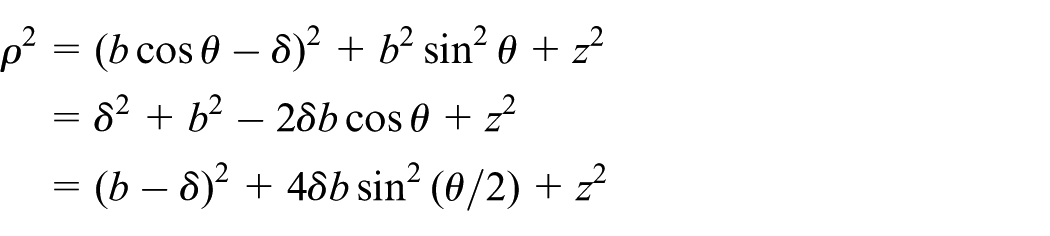
so that, the following integral must be evaluated

By defining

Now on making the further substitution

where

Note that in terms of the usual parameters of the hypergeometric function where
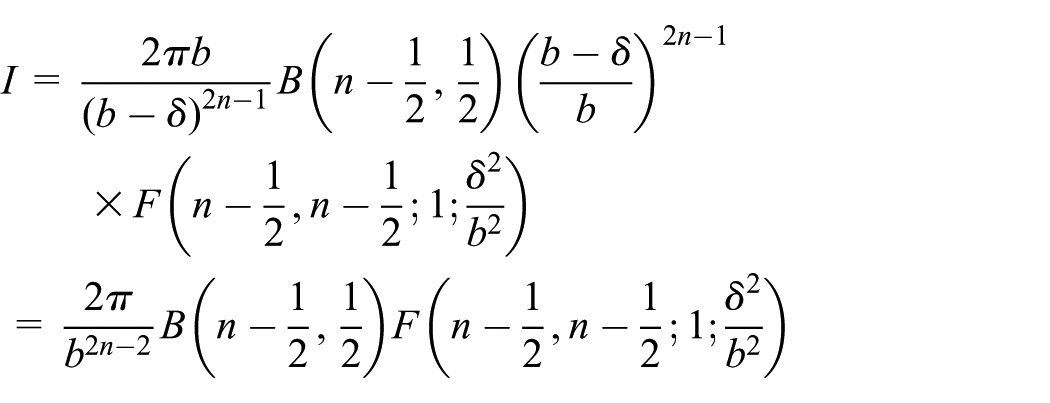
Next, the total interaction of an atomic point
The same cylinder defined in cylindrical coordinates by

In a similar manner to that described above, the integral I is of the following form

whereupon on again employing the quadratic transformation

Some important mathematical formulae are derived which may be exploited to calculate the interaction energy between two nanostructures. Analytical expressions for an atomic point (i.e. a single atom) with various shaped molecules have been determined. In more complicated atomic configurations involving two or more molecules, another surface integral of the atomic point must be evaluated to determine the total interaction energy of the system. In the following sections, a number of nanotechnology applications are surveyed which have exploited these formulae to determine the properties of the systems.
Mechanics of nanostructures
Nanostructures such as carbon nanotubes, nanopeapods, nanocones, and carbon onions exhibit outstanding physical and mechanical properties such as their high strength, high flexibility, and low weight, and they provide a basis for the creation of many novel nano-devices. One particular application which has attracted much attention is the nano-oscillator,12,37,95,96 which is able to generate frequencies in the gigahertz range,
12
and which may form the basis of a number of ultrahigh-frequency devices in the computer industry. Since the discovery of ultra low friction by Cumings and Zettl,
95
double-walled carbon nanotube oscillators have been widely studied using both molecular dynamics simulations and experiments.12,13,96–98 In addition, carbon nanotubes have received much attention for medical applications, especially their use as nanocontainers for drug and gene delivery. In particular, a well-known self-assembled hybrid carbon nanostructure, so-called nanopeapods, may be regarded as a model for possible drug carriers, where the carbon nanotube can be thought of as the nanocontainer, and the
The Lennard-Jones potential together with the continuous approximation has been successfully employed in a number of studies to determine the van der Waals interaction energy and the force between two interacting non-bonded nanostructures. In particular, several authors determine the molecular interaction between a fullerene and carbon nanotubes.8,36–46 Girifalco
36
determines the interaction energy between two
Cox et al.38,39 have proposed the important notions of suction and acceptance energies for the encapsulation behavior of an atom and a
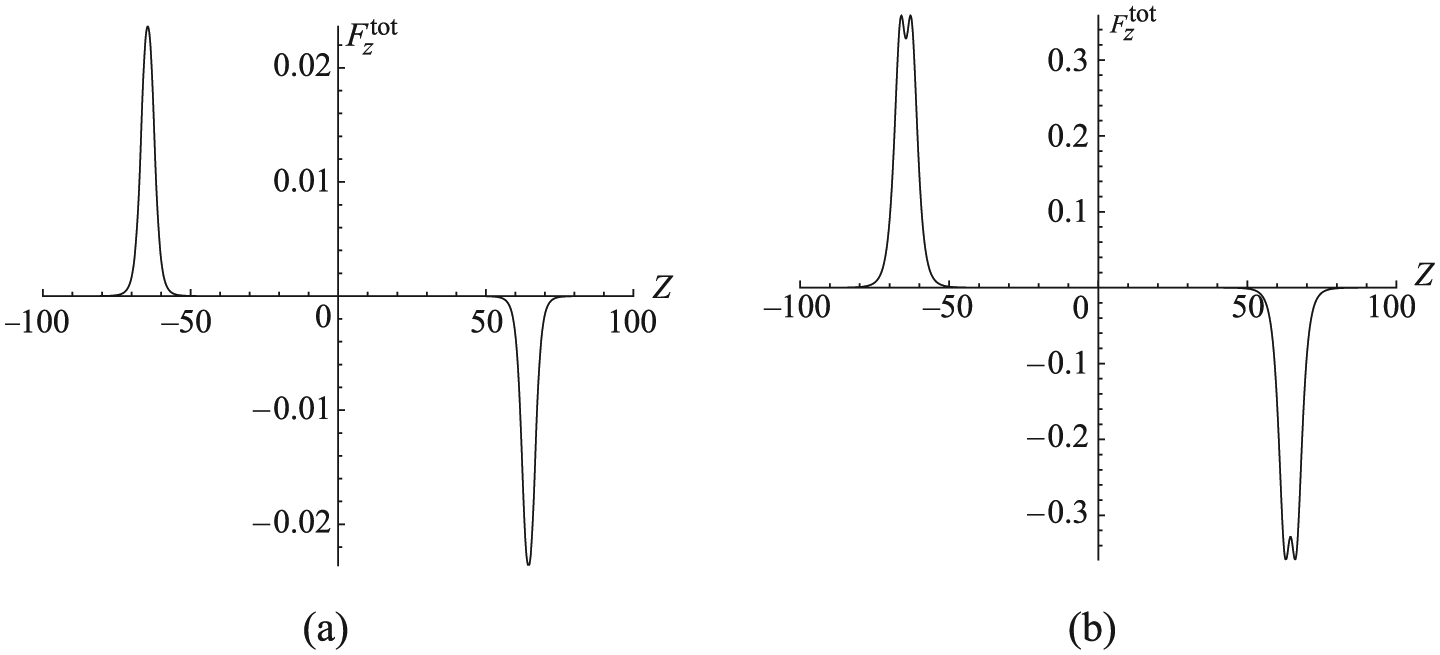
Plot of forces for (a) atom oscillating inside (6, 6) carbon nanotube and (b)
Cox et al.40,43,44 also study the mechanics of spherical and spheroidal fullerenes entering carbon nanotubes. Particularly, Figure 4 shows the energy profiles for spheroidal
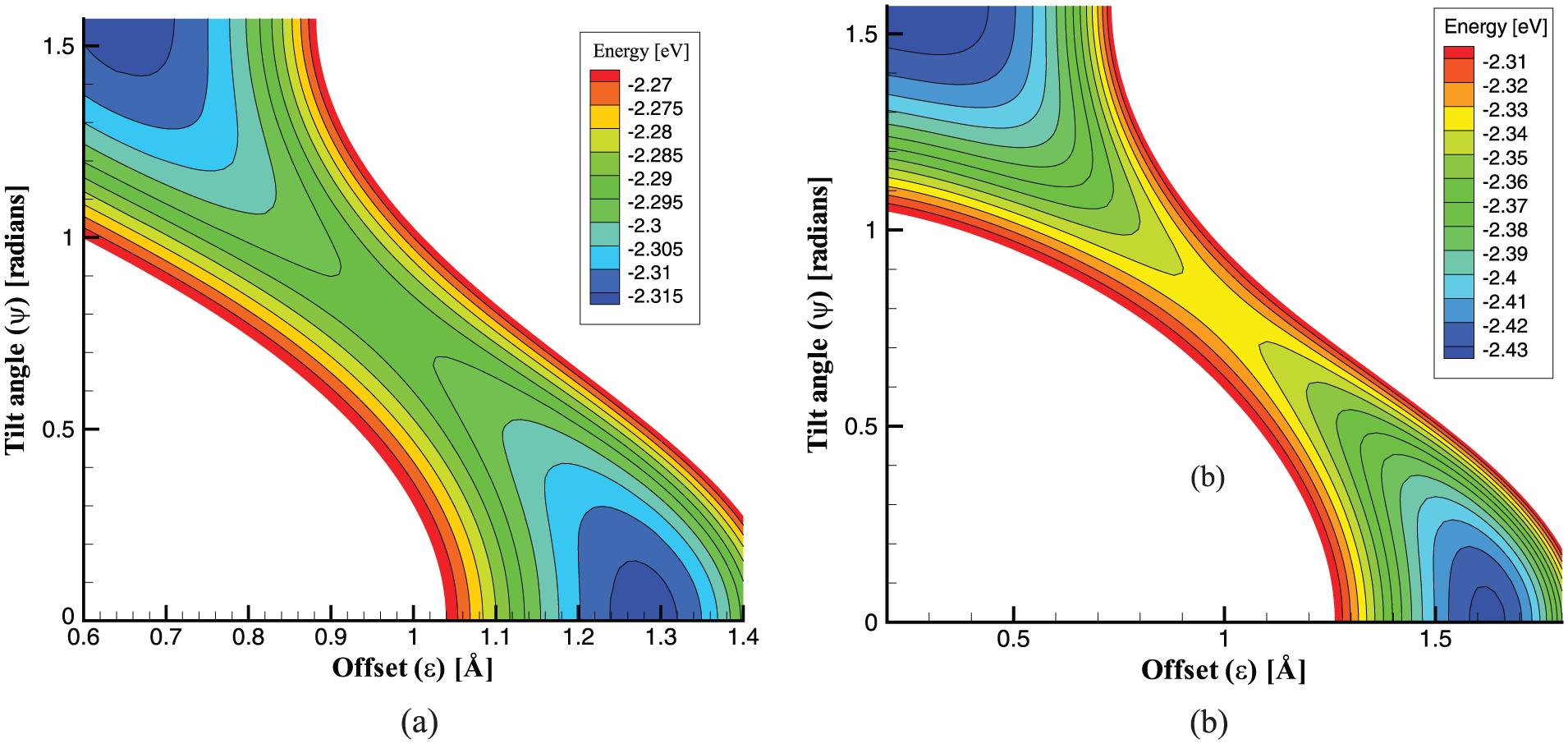
Contour plot of interaction energy for (a)
For two concentric cylindrical carbon nanotubes, Zheng and Jiang 12 determine the van der Waals restoring force between the inner and outer shells of a multi-walled carbon nanotube and subsequently predict a gigahertz frequency of the oscillatory motion. Baowan and colleagues14,15 determine analytical expressions for the suction energy and offset configurations of double-walled carbon nanotubes and also predict the gigahertz frequency for the nanotube oscillators. A similar approach has been adopted by Cox 16 to model the behavior of forced double-walled nanotube oscillators. Ansari and colleagues18–20 consider the effects of geometrical parameters on the force distributions for the oscillatory behavior of double-walled carbon nanotubes. The effect of capped ends of double-walled carbon nanotubes is also studied by Baowan, 17 and the effect of tube radii is investigated by Tiangtrong and Baowan. 21
Ruoff and Hickman
103
consider the interaction between a spherical fullerene and a graphite sheet. For spherical carbon onions
Henrard et al. 107 use a similar technique to that proposed by Girifalco 36 and obtain the potential for bundles of single-walled carbon nanotubes. Cox and colleagues22–24 study extensively the mechanics of carbon atoms and nanotubes oscillating in carbon nanotube bundles and again utilizing the Lennard-Jones potential together with the continuous approximation, and the results obtained can be used to predict the oscillator bundle configuration which optimizes the suction energy and therefore leads to the maximum frequency oscillator.
The equilibrium configurations of carbon atoms and
MOFs and gas storage
MOFs comprise metal atoms or clusters that are linked periodically by organic molecules to establish an array such that each atom forms part of an internal surface. MOFs have delivered the highest surface areas and hydrogen storage capacities for any physisorbent and are shown to be the most practically promising material for gas storage. 113 Exposed metal sites114,115 pore sizes, 116 and ligand chemistries117,118 have been found to be the most effective routes for increasing the hydrogen enthalpy of adsorption within MOFs. The MOF adsorbent that presently holds the record for gravimetric hydrogen storage capacity at room temperature is the first structurally characterized beryllium-based framework, Be-BTB (benzene tribenzoate). Be-BTB has a Brunauer–Emmett–Teller (BET) 119 surface area of 4400 m2g−1 and can adsorb 2.3 wt% hydrogen at 298 K and 100 bar. 120 We refer the reader to Furukawa et al. 121 for a comprehensive review of the chemistry and the applications of MOFs.
The so-called Topologically Integrated Mathematical Thermodynamic Adsorption Model (TIMTAM), as proposed by Thornton and colleagues,81,82 assumes the ideal building blocks described in the section on analytical expressions for idealized molecular building blocks to represent the cavity of the structure, and then, these expressions are exploited to calculate the potential energy interactions between the gas and the adsorbate. The major advantage of the TIMTAM approach is that it provides analytical formulae that are computationally instantaneous, and therefore, many distinct scenarios can be rapidly investigated which evidently serves to accelerate material design. 81 A schematic representation for MgC60@MOF for a MOF cavity impregnated with magnesium-decorated C60 is shown in Figure 5, where the TIMTAM model is utilized to determine the energy level in the cavity for the magnesium atom. 81 Moreover, the same approach has also been proved as a useful technique to investigate the effect of pore size in MOFs.80–82,86,87
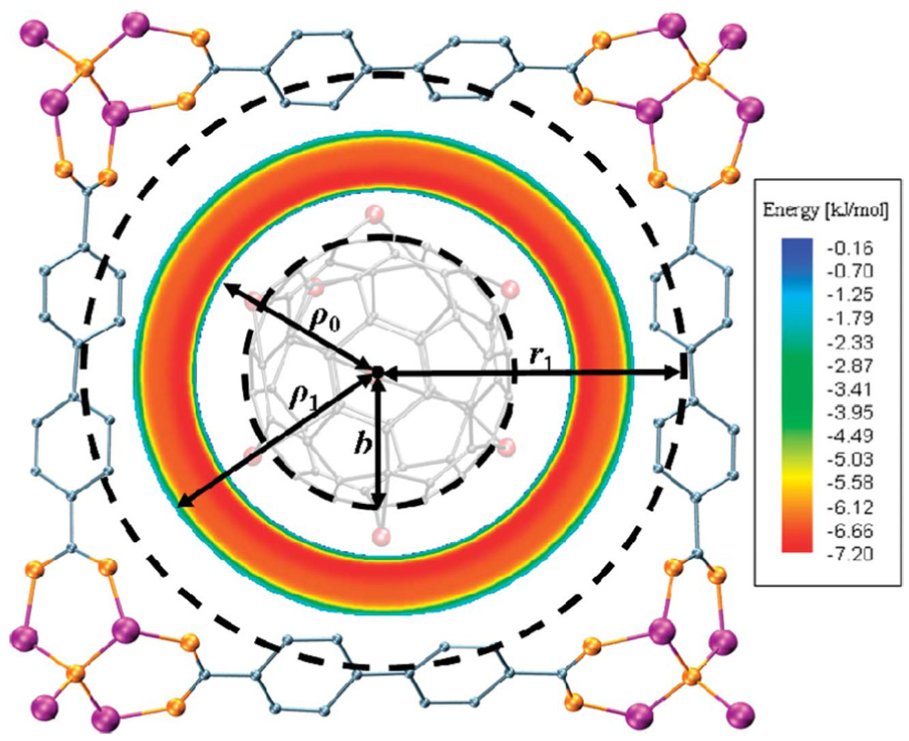
Model for
Furthermore, Chan and Hill 84 investigate the storage of hydrogen molecules inside graphene-oxide frameworks comprising two parallel graphenes rigidly separated by perpendicular ligands. These authors find 6.33 wt% for GOF-28 at a temperature of 77 K and a pressure of 1 bar which is consistent with several experimental and other computational results.122–124 Based on the assumption of no steric hindrance and a small electronic barrier, Chan and Hill 83 model the interaction of a rigidly suspended benzene molecule within a MOF, which is then used as a building block in more complex MOFs.
For the specific gas molecule, benzene, Tran-Duc and colleagues65–69 extensively investigate the equilibrium configuration of benzene dimer adsorbed on graphene sheet,
Molecular selective and separation
Water molecule has a simple chemical structure and is often a basic unit in many biomolecules. The determination of water separation can be envisaged as the first step in a study of the selective separation of more complicated molecules. Hilder and Hill 59 determine the maximum velocity for a single water molecule entering a carbon nanotube, and their model predicts that the radius of the carbon nanotube must be at least 3.464 Å for acceptance of a water molecule, and that a radius of 3.95 Å provides the maximum uptake or suction energy. Chan and Hill 61 utilize the same mathematical technique to investigate the transport of water through carbon nanotubes and suggest that their results rapidly reduce the computational time for the full numerical calculation. As an alternative for molecular selectivity, Garalleh et al.62–64 determine the interaction energy between water and various other biomolecules.
Ions are atoms or molecules in which the total number of electrons does not equal the total number of protons, giving the atom a net positive or negative electrical charge. On using the applied mathematical approach, Chan and Hill75,76 investigate the interaction energy between various types of atoms and ions, namely, Mn2+, Au, Pt, Na1+, and Li1+ on graphene sheet. These authors determine the equilibrium position for the atom/ion on the surface of the graphene sheet and the minimum intermolecular spacing between two graphene sheets. Furthermore, carbon nanotubes have been used to facilitate the transport and separation of atoms and ions.77,78 Similarly, Rahmat et al.
79
determine the suction and the acceptance behavior of
The selective separation of biomolecules is a critical process in food, biomedical, and pharmaceutical industries. Baowan and Thamwattana 50 utilize the Lennard-Jones potential function and the continuous approximation to separate trypsin and lysozyme using mesoporous silica. These authors predict that the silica pores with radii lying in the range 17.23 and 21.24 Å will only allow lysozyme to be encapsulated. Using the same approach, Thamwattana et al. 51 investigate three model configurations for bovine serum albumin to be encapsulated inside carbon nanotubes, as indicated in Figure 6. They conclude that a critical radius of pore or tube is crucial for the design to facilitate maximum loading of proteins and drug molecules.

Three possible configurations for bovine serum albumin encapsulated in a carbon nanotube and modeled: (a) as three-connected spheres, (b) as a prolate-ellipsoid, and (c) as a right-cylinder.
The far more complicated biomolecule DNA has been examined by Alshehri et al.,52–55 who determine equilibrium positions of a DNA strand absorbed onto a graphene sheet or encapsulated inside a carbon nanotube. These authors find that a space of approximately 20 Å is required for the absorption of DNA onto the graphene sheet.
53
Moreover, they observe that double-stranded DNA is encapsulated inside a single-walled carbon nanotube of radius larger than 12.30 Å, and they show that the optimal radius of the single-walled carbon nanotube to enclose a double-stranded DNA is 12.8 Å.52,54,55 Furthermore, since lipid bilayers, lipid nanotubes, and liposomes are potential candidates for use in molecular separation, Baowan et al.
56
determine the penetration and encapsulation of
Targeted drug delivery
The prospect that nanocapsules may realize the “magic bullet” concept, as first proposed at the beginning of the 20th century by the Nobel Prize winner Paul Ehrlich (1854–1915), generated immense interest in their development. The ideal drug carrier, or “magic bullet,” is envisaged as a transporter of drugs or other molecular cargo to a specific site in the body which then unloads the cargo in a controlled manner. Although this notion may sound like science fiction, the advent of nanotechnology means that it is rapidly becoming scientific fact. Despite the prominence of carbon nanotubes in the broader area of nanotechnology, the field of nanotube biotechnology is in its infancy, and there is still much work that needs to be accomplished before specific products can be produced. Drug delivery is one of the most promising biomedical applications of nanotechnology, and as stated by Hillebrenner et al. 125 in a review of template-synthesized nanotubes for biomedical delivery applications, “The future challenges for nanotubes as drug delivery vehicles are substantial but not insurmountable.”
Again following the Lennard-Jones potential function and the continuous approximation approach, Hilder and Hill determine the energy behavior and suction characteristics in the encapsulation of various drugs into nanotubes including cisplatin,126,127 paclitaxel, and doxorubicin 128 and into other nanotubes such as boron nitride, silicon, and boron carbide. 129 Their results predict an appropriate tube size that gives rise to the optimum encapsulation mechanics for a prescibed drug molecule. Moreover, Hilder and Hill 130 also examine nanosyringes comprising double-walled carbon nanotubes to inject DNA or anticancer drugs directly into the cell. The reader is referred to Hilder and Hill 131 for a comprehensive review on the various models for drug release using a nanotube carrier, as indicated in Figure 7. Ansari et al. 132 employ both a hybrid discrete–continuum model and molecular dynamics simulation to study the offset of cisplatin in a single-walled carbon nanotube and conclude that the methods give comparable results.

Outline of proposed drug delivery process: (a) nanotube surface is functionalized with a chemical receptor and drug molecules are encapsulated, (b) open end is capped, (c) nanocapsule is ingested and locates to target site due to functionalized surface, (d) cell internalizes capsule, for example, by receptor-mediated endocytosis, (e) cap is removed or biodegrades inside cell, and (f) drug molecules are released.
Summary
This review has focused on the use of applied mathematical modeling to determine the mechanical energy behavior of nanostructures. We have concentrated on those applications for which the 6-12 Lennard-Jones potential energy function and the continuous approximation apply. The continuous approximation assumes that the total molecular interaction energy of the system involving a discrete atomic configuration can be approximated by a uniform distribution of atoms, either throughout a region or over a bounding surface. First, analytical expressions for various molecular building blocks are evaluated to provide the major contribution to the interaction energy of the actual complicated atomic configuration. The principal mechanical properties of the nanostructure can then be determined from the energy distribution, which gives rise to the force distribution, equilibrium configurations, and oscillatory behavior. Moreover, this applied mathematical approach has been illustrated to determine the gas storage characteristics of MOFs. The same approach has also been successfully applied to both organic and inorganic molecules, including systems of biomolecules involving protein and enzyme selective separation and targeted drug delivery. In summary, although this robust mathematical approach has already been successfully exploited in many applications, its most important future role will likely be either as a component or as the first iteration in large-scale computational calculations to determine the molecular interaction energy of complex atomic configurations, and such use will significantly increase computational efficiency.
Footnotes
Acknowledgements
The authors gratefully acknowledge the Endeavour Research Scheme for the provision of two postdoctoral fellowships for D.B. D.B. is also grateful for the support of the Thailand Research Fund (RSA5880003). The authors also wish to acknowledge all their numerous colleagues involved in this work.
Academic Editor: Michal Kuciej
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Much of the research was supported by the Australian Research Council.