
Abstract
Elucidation of the molecular mechanisms involving the initiation and progression of radiation-induced esophageal injury (RIEI) is important for prevention and treatment. Despite ongoing advances, the underlying mechanisms controlling RIEI remain largely unknown. In the present study, RNA-seq was performed to characterize mRNA profiles of the irradiated rat esophagus exposed to 0, 25, or 35 Gy irradiation. Bioinformatics analyses including dose-dependent differentially expressed genes (DEGs), Gene Ontology (GO), Kyoto Encyclopedia of Gene and Genome (KEGG) pathway, protein-protein interaction (PPI) network, and immune infiltration were performed. 134 DEGs were screened out with a dose-dependent manner (35 Gy > 25 Gy > control, or 35 Gy < 25 Gy < control). GO and KEGG analyses showed that the most significant mechanism was IL-17 signaling-mediated inflammatory response. 5 hub genes, Ccl11, Cxcl3, Il17a, S100a8, and S100a9, were identified through the intersection of the DEGs involved in inflammatory response, IL-17 pathway, and PPI network. Additionally, immune infiltration analysis showed the activation of macrophages, monocytes, T cells, NKT cells, and neutrophils, among which macrophages, monocytes, and neutrophils might be the main sources of S100a8 and S100a9. Thus, these findings further our understanding on the molecular biology of RIEI and may help develop more effective therapeutic strategies.
Introduction
Radiation-induced esophageal injury (RIEI) is a common complication of radiotherapy (RT), particularly in patients with lung cancer, esophageal cancer, and mediastinal tumor. 1 Esophagitis is an early clinical symptom of RIEI, and severe esophagitis can greatly prolong the treatment period through additional hospitalization, and clinical symptoms such as esophageal ulcer, dysphagia, odynophagia, and retrosternal pain. Late-onset RIEI includes esophageal stricture, sclerosis, and tracheoesophageal fistula. 2 The occurrence of these toxicities is a serious challenge for radiation oncologists and leads to the delay or interruption of RT treatment, which seriously affects the tumor control probability, long-term survival, and patients’ life quality. 3 An analysis of the radiation therapy oncology group (RTOG) database showed that 95% patients with locally advanced non–small-cell lung cancer developed different grade of esophagitis, and 33% experienced severe esophagitis (Grade ≥3), peaking within the first or second month of RT. 4 Currently, some agents have been reported to demonstrate a certain therapeutic effect for radiation-induced esophagitis, including EGCG, 5 amifostine, 6 glutamine, 7 manganese superoxide dismutase (MnSOD) plasmid/liposome (PL) gene treatment, 8 GS-nitroxide (JP4–039), 9 indomethacin, 10 and sucralfate. 11 However, the effective prevention and treatment for RIEI still remains challenging. Thus, it is imperative to identify novel therapeutic targets in order to develop more effective therapy for RIEI patients.
Radiation damage to the cell can be caused by the direct effect, indirect effect and bystander effect. In the direct effect, radiation hits the DNA and/or protein directly, disrupting the molecular structure. 12 In the indirect effect, radiation hits the water and other organic molecules, producing free radicals and reactive oxygen species (ROS). 12 In the bystander effect, non-irradiated cells exhibit the biological response to signals transmitted by irradiated cells, resulting in toxic effects on adjacent non-irradiated tissues. 13 Despite these three effects mentioned above, the underlying mechanisms involved in the development of RIEI remain unclear. Previous studies reported that free radical (e.g., ROS), inflammatory mediator (e.g., transforming growth factor-β 1, tumor necrosis factor-α, interferon-γ, interleukin-1, interleukin-18), and growth factor (e.g., epidermal growth factor, hepatocyte growth factor) were involved in the initiation and development of RIEI.14-17 Subsequently, RNA-seq dataset from Sun et al and Luo et al provided the expression profile of mRNA, lncRNA, circRNA, and miRNA in rat esophageal tissues exposed to 0 and 20 Gy irradiation.18,19 Sun et al 18 reported that 853 mRNAs and 46 lncRNAs were differentially expressed in the irradiated and non-irradiated rat esophageal tissues, which were enriched in steroid biosynthesis, tumor necrosis factor signaling pathway, focal adhesion, pathological immune responses, etc. Luo et al 19 reported that 27 miRNAs and 197 circRNAs had different expression levels, involving lipid biosynthesis, cell proliferation, cell migration, cell differentiation, focal adhesion, and sphingolipid metabolism. Although Sun et al and Luo et al advanced our understanding of molecular signaling events related to RIEI, their RNA-Seq experiments were performed on only one sample, which limited the power of detecting differentially expressed RNAs.
To gain insight into molecular basis of RIEI, it is necessary to conduct mRNA expression profiles and corresponding comprehensive bioinformatics analyses in RIEI rat model with different irradiation dosage. Thus, in the present study, rat esophageal tissues exposed to 0, 25 and 35 Gy irradiation were used in mRNA-seq experiments. Esophageal tissues collected from three rats were combined to yield one pool sample. Each group contained three pool samples. Using dose-dependent genes as an entry point, we identified key genes, biological processes and pathways that were closely associated with RIEI. Based on gene expression profile, we also estimated which immune cells were involved in RIEI. These results would inform and extend current understanding regarding the molecular mechanisms of RIEI, and may help identify biomarkers to develop novel therapeutic targets for RIEI patients.
Methods
Animal Studies
Male Sprague–Dawley (SD) rats (5 weeks old) in our study were purchased from Chengdu Dossy Experimental Animals Co., Ltd. And all experimental animal protocols had been approved by the Animal Experimentation Ethics Committee of the Second Affiliated Hospital of Chengdu Medical College (China National Nuclear Corporation 416 Hospital, Chengdu, China). We randomly assigned 27 rats to three groups (0, 25 or 35 Gy; n = 9). For stable irradiation, rats were anesthetized with intraperitoneal injections of 10% chloral hydrate, and the fur on the cervicothoracic was shaved off. During the radiation exposure, the irradiated region of esophagus was kept in the range of 3 cm × 4 cm from the lower edge of ear, and other tissues were protected by 3 cm-thick leads. Using the X-RAD 225 Irradiator (Precision X-ray (PXi), Inc., North Branford, CT), different irradiation dosage (0, 25 or 35 Gy) was administered to the esophageal region at a rate of 400 cGy/min. Weight, food intake, and water intake of rats were measured daily from the first day (d1) to the seventh day (d7).
Sample Preparation for Hematoxylin and Eosin Staining, and mRNA-Seq
Rats were sacrificed 7 days after radiation and esophageal tissues were collected. For quality assurance, the esophagus was cut into three parts and numbered. The upper part of esophagus was numbered as No.1 and was used for Hematoxylin and Eosin (HE) stanning, the middle part of esophagus was numbered as No.2 and was used for mRNA-seq, the lower part of esophagus was numbered as No.3 and was saved in liquid nitrogen. To minimize intra-group differences, esophageal structure of all rats was tested with HE staining.
Hematoxylin and Eosin Staining
Esophageal tissues of rats were fixed in paraformaldehyde (4%), and subsequently embedded perpendicularly in paraffin. Then 3 μm-thick paraffin sections were deparaffinized and stained with HE. Under the light microscope (Olympus BX53, Tokyo, Japan), the morphology changes of esophageal tissues were observed clearly.
mRNA-seq
Esophageal tissues collected from three rats were combined yielding one pool sample. Each group (0, 25 or 35 Gy) contained three pool samples. Total RNA was extracted from nine pool samples using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA). RNA integrity was evaluated with Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and only the samples with RNA Integrity Number (RIN) ≥ 7 were applied to the following analysis. The libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA). Then these libraries were sequenced on the Illumina sequencing platform (HiSeqTM 2500). Raw data (raw reads) were processed using Trimmomatic. 20 To filter out the clean reads, the low-quality reads and the reads containing ploy N were considered as useless reads. Then, the clean reads were calculated to reference genome using hisat2. 21 FPKM value of each gene was obtained by cufflinks 22 and the read counts of each gene were counted by htseq-count. 23 The processed data consisted of gene ID, gene symbol, gene description, FPKM value, and read count. Principal Component Analysis (PCA) was performed to visualize the gene expression pattern by the FactoMineR and factoextra R packages. The differentially expressed genes (DEGs) were identified using the DESeq R package. The threshold of DEGs was FDR <.01 and |log (fold change) | ≥1.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Pathway Analyses
The gene symbols were imported into Metascape database (http://metascape.org/), 24 and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed. And GO analysis comprised three terms: biological process (BP), molecular function (MF), and cellular component (CC). Terms with a P-value < .05, a minimum count of 3 and an enrichment factor >1.5 were considered significant.
Construction of Protein-Protein Interaction Network
The gene symbols were fed into the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/) to construct a functional protein-protein interaction (PPI) network. The interactions with the composite score >.4 were considered statistically significant. Then, the PPI network was visualized by Cytoscape software. The node genes were screened out with the cut-off criteria of connectivity degree ≥3.
Immune Cell Infiltration Analysis
ImmuCellAI-mouse (http://bioinfo.life.hust.edu.cn/ImmuCellAI-mouse/#!/) is a tool to estimate the abundance of 36 immune cells based on gene expression profile from RNA-Seq or microarray data. 25 The 36 cell types consist of three layers: (1) the 7 major immune cell types (B cells, monocytes, macrophages, T cells, dendritic cells (DCs), NK cells, and granulocytes), (2) the 6 subtypes of B cells (B1, follicular B, germinal center B, marginal zone B, memory B, and plasma cells), the 4 subtypes of T cells (CD4 T, CD8 T, NKT, and γσT cells), the 2 subtypes of macrophages (M1 macrophage and M1 macrophage), the 4 subtypes of DCs (cDC1, cDC2, MoDC, and pDC), and the 4 subtypes of granulocytes (basophil, eosinophil, mast cell, and neutrophil), (3) the 9 subtypes of CD4 T and CD8 T cells (CD4+ naïve, CD4+ memory, Treg, T helper, CD8+ naïve, CD8+ central memory, CD8+ effector memory, cytotoxic and exhausted cells). The corresponding gene set was converted to mouse homologs using the Msigdbr R package. Then, the transformed gene expression profile was uploaded to ImmuCellAI-mouse to estimate the abundance of immune cells. The differences of immune cell infiltration among control group, 25 Gy group and 35 group were tested by Tukey’s multiple comparisons test. Significance was noted by P-value: *P < .05; **P < .01; ***P < .001, and ****P < .0001.
Results
The Alteration of Radiation-Induced Esophageal Injury in a Rat Model
As we reported previously,
26
esophageal irradiation with dose ≥25 Gy X-ray caused morphological and functional damage to the esophagus in a rat model. To ensure the quality of all esophageal tissues for mRNA-seq, we detected the alteration of body weight, food intake, water intake, and esophageal structure in all three groups of rats (0, 25 or 35 Gy; n = 9). As shown in Figures 1A, 1B, and 1C, 25 or 35 Gy-irradiated rats showed a significant and gradual decrease in body weight, food intake, and water intake from the first to seventh day after irradiation. And there were no significant differences in body weight, food intake, and water intake between 35 Gy and 25 Gy groups. The rats in 0 Gy group were active and restless, with smooth and shiny fur, whereas 25 or 35 Gy-irradiated rats displayed a weak state, decreased activity and increased yellow hair and decreased luster (Figure 1(D)). Meanwhile, HE staining was performed on the esophagus of rats 7 days post irradiation. The results showed that the 0 Gy group had intact and healthy esophageal structure, whereas 25 or 35 Gy-irradiated esophagus presented obvious pathological changes: epithelial thickening (25 Gy), epithelial shedding (35 Gy), inflammatory cell infiltration (25 or 35 Gy) (Figure 1(E)). And the esophageal damage of 35 Gy group was more serious than that of 25 Gy group. Based on the above results, the quality of all esophageal tissues was ensured from the perspective of phenotype. The alteration of radiation-induced esophageal injury in a rat model. A single dose of 0, 25, or 35 Gy X-ray was administered to the esophageal area (n = 9). (A) Average daily weight of each group. (B) Average daily food intake of each group. (C) Average daily water intake of each group. (D) General appearance of each group. (E) HE staining of each group.
Analysis of Differentially Expressed Genes and Dose-Dependent Genes
mRNA-seq was performed on the irradiated and non-irradiated rat esophageal tissues, which were collected 7 days after irradiation with 0 Gy (control group), 25 Gy (25 Gy group), or 35 Gy (35 Gy group). After processing the raw data, gene expression profiling was obtained, including gene ID, gene symbol, gene description, FPKM value, and read count of each gene. First, we performed a principal component analysis (PCA) on gene expression profile of all the samples. Each point on the PCA plot represented the gene expression profile of an individual sample. As shown in Figure 2(A), there was a significant degree of separation among the three groups, reflecting obvious differences in gene expression. Next, analysis of differentially expressed genes (DEGs) was carried out on the three pairwise comparisons, including 25 Gy Vs control, 35 Gy Vs control, and 35 Gy Vs 25 Gy. Using FDR <.01 and |log (fold change) | ≥1 as the cut-off criterion, we observed that 432 up-regulated and 987 down-regulated DEGs between 25 Gy and control groups, 2606 up-regulated and 2646 down-regulated DEGs between 35 Gy and control groups, 1242 up-regulated and 1076 down-regulated DEGs between 35 Gy and 25 Gy groups (Figure 2(B)). These above results suggested that irradiated rat esophageal tissue obtained more DEGs with the increase of radiation dose. Analysis of differentially expressed genes and dose-dependent genes. (A) The degree of separation among the three groups of samples in the PCA score map. (B) Number of up- or down-regulated genes among the three pairwise comparisons. (C) Venn diagram showed the respective frequency of up-regulated and down-regulated genes among the three pairwise comparisons, the red circle represents dose-dependent genes of 35 Gy > 25 Gy > Control, or 35 Gy < 25 Gy < Control. (D) Heatmap plot of dose-dependent genes among the three groups.
To explore which DEGs presented the dose-dependent change, up-regulated and down-regulated DEGs were respectively selected according to the cut-off criterion of radiation doses (35 Gy > 25 Gy > control, or 35 Gy < 25 Gy < control). 67 DEGs were up-regulated with a change of 35 Gy > 25 Gy > control, and 67 DEGs showed a down-regulated change of 35 Gy < 25 Gy < control (Figure 2(C) and 2(D)). Thus, these 134 genes were considered as dose-dependent genes, which may play a vital role in the progression of RIEI.
Investigation of the Potential Functions and Pathways
To elucidate the potential biological functions and pathways of these dose-dependent genes, Gene Ontology (GO) and KEGG pathway analyses were performed on the above 134 DEGs. Based on Metascape database, we observed a total of 328 Gene Ontology assignments, including 279 BPs, 41 MFs, and 8 CCs. In the BP category, most genes were involved in inflammatory response, leukocyte migration, granulocyte migration, antimicrobial humoral response, and humoral immune response. In the MF category, most genes may play roles in signaling receptor regulator activity, sialic acid binding, SH2 domain binding, receptor ligand activity, and calcium-dependent protein binding. In the CC category, a large percentage of the genes were associated with spindle microtubule, phagocytic vesicle, endocytic vesicle, cytoplasmic side of membrane and cytoplasmic side of plasma membrane. The top 10 significant terms of GO classification were illustrated in Figure 3. According to the P-value of GO enrichment analysis, inflammation-related responses were the most significantly enriched terms, which was consistent with pathological phenotype. Gene ontology (GO) analysis of dose-dependent genes. GO analysis comprised three terms: biological process, molecular function, and cellular component. The top 10 significant GO terms were shown.
KEGG Pathway Analyses of the Dose-Dependent DEGs.

Identification of the Most Significant Pathway and Hub Genes
To identify the most significant dose-dependent genes “hub genes” associated with RIEI, we performed a PPI network construction of the above 134 DEGs on the basis of the STRING database. Finally, 64 nodes and 100 edges were present in the PPI network complex (Figure 4(A)), in which 29 node genes were selected with the cut-off criteria of connectivity degree ≥3 (Figure 4(B)). Subsequently, GO biological process and KEGG pathway analyses of these 29 node genes were further carried out (Table2). The node genes were involved in the following top 10 biological processes: inflammatory response, leukocyte migration, innate immune response, humoral immune response, granulocyte migration, neutrophil chemotaxis, granulocyte chemotaxis, neutrophil migration, myeloid leukocyte migration, and response to lipopolysaccharide. And the top 10 significantly enriched pathways were IL-17 signaling pathway, Staphylococcus aureus infection, NF-kappa B signaling pathway, natural killer cell mediated cytotoxicity, TNF signaling pathway, leukocyte transendothelial migration, osteoclast differentiation, malaria, NOD-like receptor signaling pathway, and AGE-RAGE signaling pathway in diabetic complications. Protein–protein interaction (PPI) network of dose-dependent genes. (A) The PPI network was visualized using Cytoscape software. The node size was proportional to the connectivity degree. The genes with no connectivity were not present in the network. Red indicates that the expression of genes is up-regulated, and blue indicates that the expression of genes is down-regulated. (B) 29 node genes were screened out with the cut-off criteria of connectivity degree ≥3 from the PPI network complex. GO Biological Process and KEGG Pathway Analyses of the Node Genes.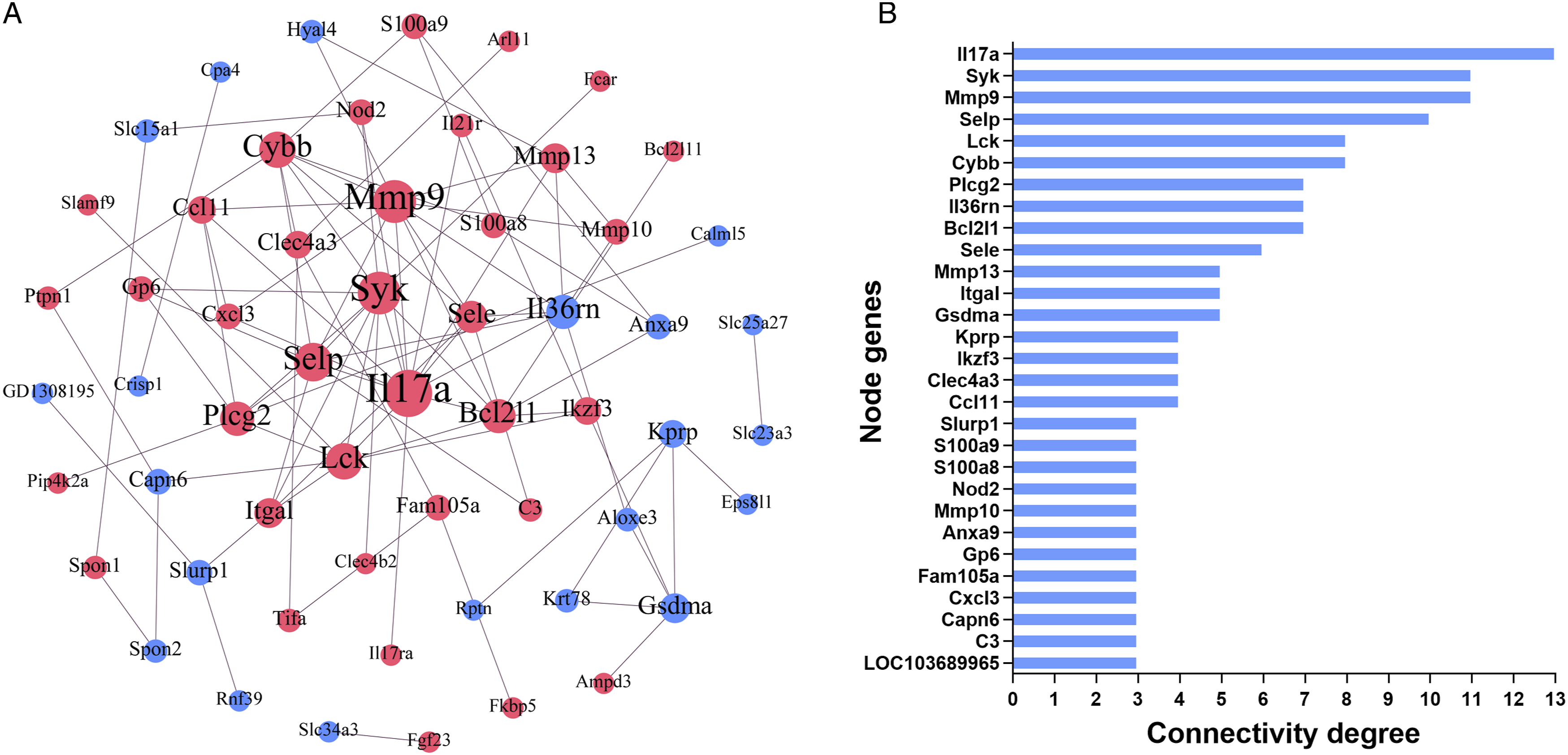

According to GO biological process and KEGG pathway analyses for the entire dose-dependent gene set and 29 node genes, we observed that the most significant biological function and pathway were inflammatory response and IL-17 signaling pathway. Therefore, the common genes present in inflammatory response and IL-17 signaling pathway were selected as hub genes, including Ccl11, Cxcl3, Il17a, S100a8, and S100a9. Based on the gene expression profile, the mRNA level alterations of Ccl11, Cxcl3, Il17a, S100a8 and S100a9 showed an obvious increase in 25 Gy and 35 Gy groups, compared with the control group (Figure 5). Among them, S100a9 presented a major increase of 225.6‐ and 2464‐fold in 25 Gy and 35 Gy groups, respectively (Supplementary Table s1). Taken together, Ccl11, Cxcl3, Il17a, S100a8, and S100a9 were likely to be involved in RIEI by IL-17 signaling pathway-mediated inflammatory response. Expression level changes of 5 hub genes among the three groups. Ccl11, Cxcl3, Il17a, S100a8, and S100a9 were identified as hub genes associated with radiation-induced esophageal injury in a rat model.
Analysis of Immune Infiltration in RIEL Rat Model
According to the above esophageal pathological changes and bioinformatics analysis, we observed that inflammation reaction was the key link of RIEI progression. To investigate which immune cells were involved in RIEI, ImmuCellAI-mouse algorithm was applied to estimate the abundance of 36 immune cells based on gene expression profile from three groups. The immune infiltration score of 35 Gy group was higher than that of 25 Gy group and control group, and there was a trend for 25 Gy group showing higher infiltration score than in control group (Supplementary Figure s1). There are 17 types of immune cells with different abundance among three groups (Figure 6). B cell, dendritic cell, plasma cell, MoDC cell, and mast cell were decreased in 25 Gy and 35 Gy groups, and there was no significant difference between 25 Gy and 35 Gy groups. Granulocyte, germinal center B, and cDC1 cell in 35 Gy group were lower than those in control group. The abundance of NK cell, eosinophil cell was decreased in 25 Gy and 35 Gy groups, and 35 Gy group was lower than 25 Gy group. The abundance of monocyte, NKT cell in 35 Gy group was higher than that in control group. T cell and neutrophil cell were increased in 25 Gy and 35 Gy groups, and there was no significant difference between 25 Gy and 35 Gy groups. Macrophage, including M1 macrophage, and M2 macrophage, was increased in 25 Gy and 35 Gy groups, and 35 Gy group was higher than 25 Gy group. As shown in Figure 6, the most conspicuous changes were noted in macrophages, which increased with the increase of radiation dose. These findings provided further evidence for understanding the mechanism of RIEI. Analysis of immune infiltration among the three groups. The abundance of 36 immune cells was estimated based on gene expression profile from three groups. The differences of immune cell infiltration among control group, 25 Gy group and 35 group were tested by Tukey’s multiple comparisons test. Significance was noted by P-value: *P < .05; **P < .01; ***P < .001, ****P < .0001.
Discussion
The advent of high-throughput technologies has greatly advanced the study of radiation damage. Based on animal models of radiation damage, researchers are generating massive amounts of omics-scale data. In the present study, using mRNA-seq as a powerful and widely adaptable analysis technique, we assessed the mRNA changes of rat esophagus exposed to different radiation doses. Further bioinformatics analysis was performed to investigate key genes, biological processes, pathways, and immune cells involved in RIEI.
We identified a total of 134 dose-dependent DEGs with a change of 35 Gy > 25 Gy > control or 35 Gy < 25 Gy < control. The enrichment analyses of these 134 DEGs showed that inflammatory response and IL-17 signaling pathway were the most significant biological function and pathway, respectively. 5 hub genes, Ccl11, Cxcl3, Il17a, S100a8, and S100a9, were identified through the intersection of the DEGs involved in inflammatory response, IL-17 signaling pathway, and PPI network construction. Immune infiltration analysis revealed the activation of macrophages, monocytes, T cells, NKT cells, and neutrophils, among which macrophages had the most conspicuous changes.
As we know, inflammatory response is one of the most important pathological mechanisms of radiation-induced normal tissue toxicity.
27
Radiation-induced inflammation is initiated by the production of free radicals, the induction of cell death, and the activation of cytokines, chemokines, and growth factors.
28
However, the inflammatory response of normal tissues to radiation is highly dependent on the radiation dose. The exposure of body cells to low dose (<1 Gy) stimulates anti-inflammatory effects, while higher dose (>2 Gy) is pro-inflammatory.29,30 In RIEI model of 5-week-old male SD rats, dose ≥25 Gy X-ray could induce an obvious infiltration of pathological immune cells in the irradiated esophagus. Multiple pathways are involved in the inflammatory response, such as nuclear factor kappa B (NF-kB), Janus kinase/signal transducers and activators of transcription (JAK-STAT), toll-like receptor (TLR) pathways, cGAS/STING, and mitogen-activated protein kinase (MAPK).
31
In our study, IL-17 signaling pathway was identified as the top inflammation-related signaling pathway associated with RIEI, in which 8 dose-dependent genes (Ccl11, Mmp9, S100a9, S100a8, Mmp13, Cxcl3, Il17a and Il17ra) were up-regulated, and 1gene (Defb4) was down-regulated. Of them, Il17a, also known as IL-17, is an upstream molecule of IL-17 signaling pathway, and is produced by a distinct subset of CD4+ T helper (Th) cells called Th17 cells.
32
The biologically active IL-17 interacts with type I cell surface receptor IL-17R (renamed IL-17RA) and activates several signal cascades, leading to the induction of chemokines.
32
These chemokines recruit the immune cells, such as monocytes and neutrophils to the site of inflammation.
32
Thus, we speculated the following process in RIEI rat model: ionizing radiation induces the expression of Il17a and Il17ra, Il17a binds to Il17ra to form the functional complex, the complex stimulates the expression of pro-inflammatory chemokines and cytokines including Ccl11, Cxcl3, S100a9, S100a8, Mmp9 and Mmp13, these chemokines and cytokines recruit macrophages, monocytes, T cells, NKT cells, and neutrophils to the irradiated esophagus (Figure 7). Schematic representation for the mechanism of radiation-induced esophageal injury in rats. Radiation induces the expression of Il17a and Il17ra, Il17a binds to Il17ra to form the functional complex, the complex stimulates the expression of pro-inflammatory chemokines and cytokines including Ccl11, Cxcl3, S100a9, S100a8, Mmp9, and Mmp13, these chemokines and cytokines recruit macrophages, monocytes, T cells, NKT cells, and neutrophils to the irradiated esophagus.
Among 5 hub genes, S100a8 and S100a9 presented the largest increase with hundreds to thousands of fold changes. S100A8 and S100A9, members of calcium-binding S100 protein family, often exist in the form of heterodimer and are mainly derived from immunocytes, such as neutrophils and macrophages. 33 S100A8/S100A9 complexes are locally released in almost all inflammatory diseases, like autoimmune diseases, rheumatoid arthritis, allergies, cardiovascular diseases, infections, and tumors, while no expression can be found in healthy tissue. 34 Therefore, S100A8 and S100A9 have been clarified as members of danger-associated molecular pattern molecules (DAMPs) family, also known as alarmins. At present, there are few studies on S100A8 and S100A9 in radiation-induced injury. Notably, two studies have reported that mRNA levels of S100a8 and S100a9 were up-regulated in irradiated skin and their expression strongly correlates with increased severity of radiation dermatitis in mice.35,36 During inflammation, neutrophils, macrophages, and monocytes strongly secrete S100A8 and S100A9 to regulate inflammatory processes by inducing inflammatory cytokines, reactive oxygen species (ROS), and nitric oxide (NO). 37 Excessive expression of S100A8 and S100A9 accelerates neutrophils and macrophages to release more cytokines, which triggers a vicious cycle and aggravates the inflammation. 38 Significantly, our results also revealed the obvious activation of macrophages, monocytes, and neutrophils in RIEI rat model, indicating that S100A8 and S100A9 actively contribute to pathological proceedings in RIEI. Additionally, multiple studies have confirmed that S100A8/A9 can serve as a candidate biomarker for diagnosis and follow-up as well as a predictor of therapeutic responses in arthritis, inflammatory bowel disease, dermatitis, vasculitis and autoimmune diseases. 39 Moreover, blockade of S100A8/S100A9 complexes represents a promising approach for local suppression of inflammatory diseases. Tasquinimod, a small-molecule inhibiting S100A8/A9 signaling, significantly ameliorated the phenotype and fibrosis of myeloproliferative neoplasm in a murine model. 40 Paquinimod, a specific inhibitor of S100A8/A9, reduces synovial activation, osteophyte formation, and cartilage damage in the osteoarthritis mouse models. 41 In a murine model of arthritis, treatment with anti-S100A9 antibodies improved the clinical score by 50%, diminished immune cell infiltration, and reduced inflammatory cytokines. 42 Therefore, these above studies and our findings suggest that the potential of S100A8 and S100A9 as the diagnostic, predictive, prognostic, and therapeutic biomarkers in RIEI.
In conclusion, we identified a substantial number of dose-dependent genes in the irradiated rat esophagus, and imputed key genes, pathways and immune cells related to Inflammation in the pathogenesis of RIEI. IL-17 signaling pathway-mediated inflammatory response may represent a new regulatory mechanism and a novel therapeutic target for RIEI. In particular, the role of S100A8 and S100A9 in RIEI should receive more scientific attention due to their properties as alarmins, but more experimental evidence is needed before its widespread application in RIEI.
Supplemental Material
Supplemental Material - Transcriptome Profiling Unveils a Critical Role of IL-17 Signaling-Mediated Inflammation in Radiation-Induced Esophageal Injury in Rats
Supplemental Material for Transcriptome Profiling Unveils a Critical Role of IL-17 Signaling-Mediated Inflammation in Radiation-Induced Esophageal Injury in Rats by Jia Yao, Jinkang Zhang, Jinlong Wang, Qian Lai, Weijun Yuan, Zhongyu Liu, Shuanghua Cheng, Yahui Feng, Zhiqiang Jiang, Yuhong Shi, Sheng Jiang, and Wenling Tu in Dose-Response
Footnotes
Author Contributions
Conceived the study: Wenling Tu, Sheng Jiang;
Drafted the manuscript: Jia Yao, Wenling Tu;
Established a rat model of RIEI: Jinlong Wang, Qian Lai, Weijun Yuan;
Performed HE staining: Shuanghua Cheng, Yahui Feng;
Performed the bioinformatics analysis: Jia Yao, Jinking Zhang, Zhongyu Liu;
Revised the manuscript: Yuhong Shi, Zhiqiang Jiang.
All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Natural Science Foundation of China (82003390 and 82103773), Medical Youth Innovation Project of Sichuan Province (Q19061), Medical Scientific Research Subject of Chengdu City (2020058), Innovation and Entrepreneurship Training Program for College Students, Chengdu Medical College (S202013705051), and the Young Talent Program of China National Nuclear Corporation.
Ethics Approval
Our study was approved by the Animal Experimentation Ethics Committee of the Second Affiliated Hospital of Chengdu Medical College (China National Nuclear Corporation 416 Hospital, Chengdu, China)
Data Availability Statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.