
Abstract
The association between regorafenib dosage in the treatment of metastatic colorectal cancer (mCRC) and efficacy is currently not well established. It was previously reported that the regorafenib dose as prescribed is associated with efficacy, but doses in actual clinical settings have not been analyzed. We retrospectively analyzed patients with mCRC who had received regorafenib as third-line or later chemotherapy between May 2013 and June 2018. Patients who were not treated in the Pharmaceutical Outpatient Clinic for compliance assessment were excluded. Overall survival was calculated using the Kaplan–Meier method. Prognostic factors, including baseline demographics and adverse events, were evaluated using Cox proportional hazard models. A total of 176 patients were enrolled. By multivariate analysis, total dose until the second cycle < 3180 mg (HR = 1.71, 95% CI, 1.20–2.44, P = .003) was one of independent negative predictors of overall survival. Median survival times of the lower-dose group (< 3180 mg) and higher-dose group (≥ 3180 mg) were 5.8 and 7.6 months, respectively (P = .045). The cumulative dose of regorafenib until the second cycle in patients with mCRC was associated with survival. It is important to individualize regorafenib dose in mCRC patients.
Introduction
Colorectal cancer is the third most commonly diagnosed cancer, with approximately 1.8 million cases worldwide in 2018. 1 The outcome of treatment has improved year by year, and the overall survival (OS) of colorectal cancer is longer than that of other cancers. However, metastatic colorectal cancer (mCRC) continues to have high mortality.
Regorafenib is an oral multitargeted kinase inhibitor that targets vascular endothelial growth factor receptor 1-3, tyrosine kinase with immunoglobulin, and epidermal growth homology domain 2, KIT, rearranged during transcription tyrosine kinase, rat fibroblastoma 1, v-raf murine sarcoma viral oncogene homolog B1, platelet-derived growth factor receptor, and fibroblast growth factor receptor. 2 In the CORRECT trial, regorafenib significantly improved survival in patients with mCRC who were refractory to standard chemotherapy. 3 Although regorafenib contributes to the survival benefit, the emergence of severe toxicities, such as hand-foot skin reaction (HFSR) and skin rash, have come to limit the continuity of regorafenib. In particular, higher frequencies of adverse events such as severe HFSR and hypertension in the Japanese subpopulation are well known compared to the non-Japanese subpopulation (HFSR: 80% vs 39.3% and hypertension: 24.6% vs 1.8%, respectively). 4 Therefore, establishment of an optimal administration method considering efficacy and tolerability is desired.
It is reported that toxicities have caused discontinuation or dose reductions in the use of regorafenib.5,6 The cumulative incidence of HFSR and liver dysfunction in a prospective observational study was higher in patients who initially received 160 mg than in those who received ≤ 120 mg. 5 As a result, dose-escalation strategies have been attempted. In the ReDOS study, in which the starting dose was 80 mg with weekly dose-escalations up to 160 mg in the dose-escalation group, a higher proportion of patients in the dose-escalation group achieved cycle 3 of treatment compared with the standard-dose group, with numerically longer OS in the dose-escalation group. 7 In the RESET study, which used another dose-escalation strategy of a starting dose of 120 mg, patients who required dose modification exhibited a better disease control rate. In addition, the study suggested that it is important in achieving disease control to continue treatment within the first 28 days. 8 These data suggest that adjustment of the regorafenib dose is important and that the cumulative dose in the early cycles may be associated with disease control. However, there are no data indicating what total dose of regorafenib would be sufficient to achieve disease control. In this study, we measured the cumulative dose of regorafenib (i.e., the actual dose taken by patients within the initial 2 cycles) and examined the relationship between the cumulative dose of regorafenib and survival in a real-world setting. The aim of this study was to examine the association between the clinical significance of the cumulative dose of regorafenib in the early cycles and treatment efficacy in patients with mCRC.
Methods
Study Population
All patients who were treated with regorafenib at the Cancer Institute Hospital between May 2013 and June 2018 were enrolled. Exclusion criteria for this retrospective study included (1) diagnosis of gastrointestinal stromal tumor, (2) enrollment in another clinical trial, (3) unclear duration of regorafenib administration because the patient transferred to another hospital, and (4) patients who were not treated in the Pharmaceutical Outpatient Clinic (POC) for compliance assessment. The clinical protocol was approved by the Institutional Review Board of the Cancer Institute Hospital (approval number 2018-1239).
Treatment
Regorafenib was administered orally as third-line or later chemotherapy. The standard dose was 160 mg/day daily for the first 21 days of a 28-day cycle. Treatment continued until disease progression, intolerable toxicity, or patient refusal. In this study, the cumulative dose until the second cycle was defined as the amount of regorafenib that patients took until day 56 because some patients experienced an irregular schedule due to delays or interruptions.
Data Collection
We gathered the following demographic data: age, gender, Eastern Cooperative Oncology Group (ECOG) performance status (PS), primary colorectal site, metastatic site (peritoneum, liver, and lung), number of metastatic sites, site of primary tumor, history of adjuvant chemotherapy, number of prior chemotherapy sessions, use of antibody drugs, regorafenib initial dose, KRAS mutations, and history of trifluridine/tipiracil (FTD/TPI) use. We collected data regarding adverse events related to regorafenib: HFSR, liver dysfunction, hypertension, skin rash, and emergency hospitalization. The severity of adverse events was evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) 4.0. 9 We evaluated the severity of HFSR as part of palmar–plantar erythrodysesthesia syndrome using NCI-CTCAE v 4.0. We retrospectively collected these data from electronic medical records. In addition, we calculated the cumulative dose of regorafenib and evaluated adherence to regorafenib using pill counts and patient-reported treatment diaries of the POC, as previously reported. 10
Statistical Analysis
OS was defined as the time from initiation of regorafenib administration to death from any cause. OS was calculated using the Kaplan–Meier method, and differences were evaluated using the log-rank test. The study population was separated into 2 groups by median regorafenib total dose until the second cycle (one group consisting of patients with total dose ≥ 3180 mg and the other with median dose < 3180 mg) in order to evaluate OS and adverse events.
Pearson’s chi-square test or Fisher’s exact test was used to compare patient characteristics and adverse events. Univariate and multivariate analyses were performed to evaluate prognostic factors using Cox proportional hazard models. We selected factors with substantial impacts (P <.2) in the univariate analysis and previously reported prognostic factors.5,11,12 The age cutoff (65 years), which is one of the prognostic factors, was based on the CORRECT study5. These were subsequently evaluated by multivariate analysis. We considered differences to be significant when the P value was < .05, and all tests were two-sided. SPSS software, version 24 (IBM Corp., Armonk, NY, USA), was used for all statistical analyses.
Results
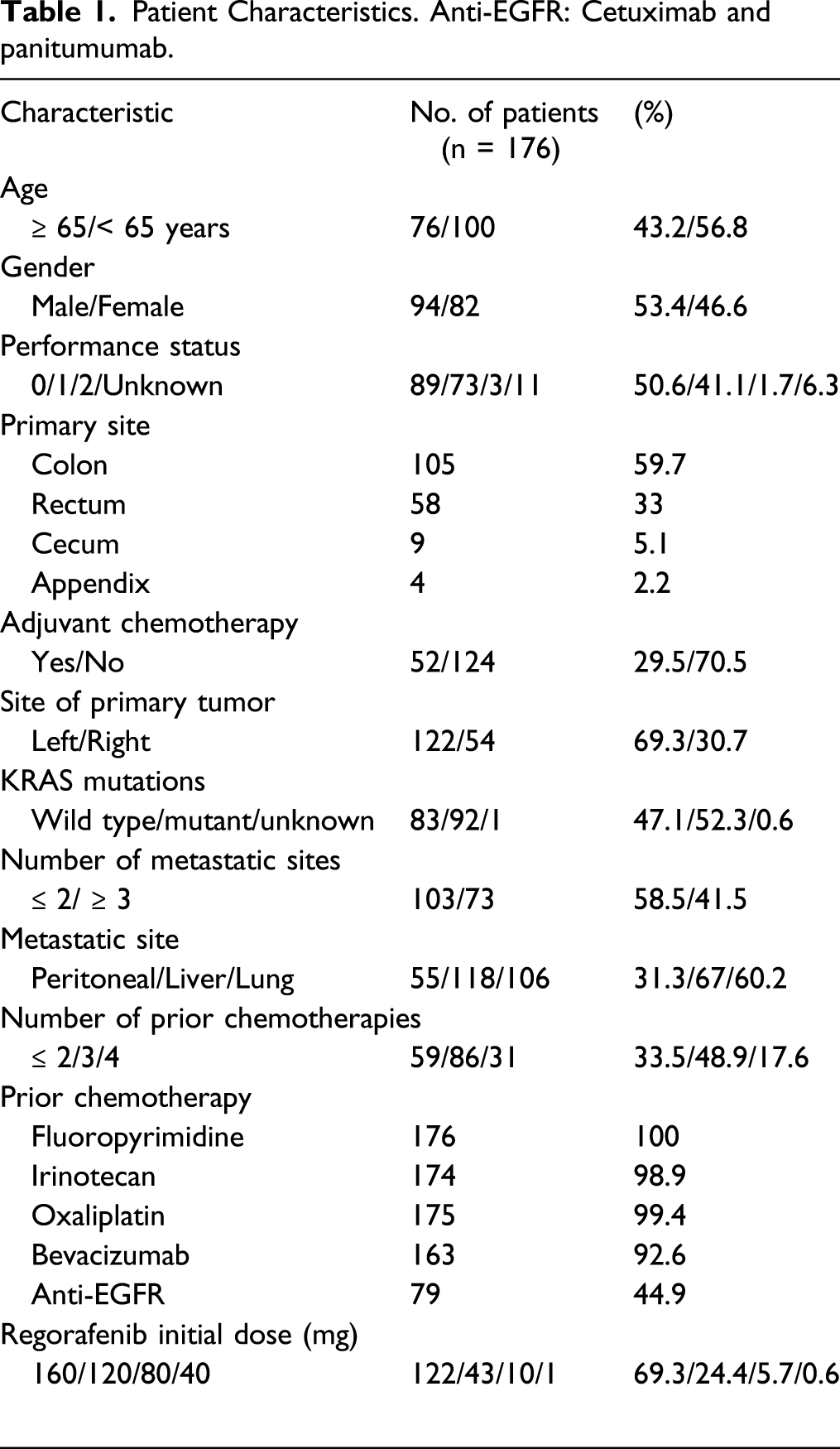
Patient Characteristics
Patient Characteristics. Anti-EGFR: Cetuximab and panitumumab.
Overall Survival and Analysis of Prognostic Factors
Multivariate Analysis of Prognostic Factors.

OS curves were probably separated according to the cumulative dose of regorafenib within the initial 2 cycles (Figure 1). Median survival times of the lower-dose group (< 3180 mg) and higher-dose group (≥ 3180 mg) were 5.8 and 7.6 months, respectively (P = .045). We also compared the patient characteristics between the 2 groups (Table 3). Gender (P = .011) and adjuvant chemotherapy (P = .023) were statistically skewed between groups. However, they were not identified as prognostic factors in the multivariate analysis. . Overall Survival Between Groups Based on Median Total Dose. Patient Characteristics Between Groups. Abbreviations: FTD/TPI, trifluridine/tipiracil. Statistical analysis: Characteristics compared by Pearson’s chi-square test (or *Fisher’s exact test)

Adverse Events Related to Regorafenib
Adverse Events Related to Regorafenib.

Statistical analysis: patient characteristics compared by Pearson’s chi-square test.
Discussion
Our multivariate analysis identified total dose until the second cycle < 3180 mg, age < 65 years, PS 2, hepatic metastasis, and regorafenib initial dose ≤120 mg as prognostic factors of regorafenib. In groups divided by median dose, regorafenib total dose was associated with OS. It should be noted that a particular cut-off value for cumulative regorafenib dose was presented because it was not reported previously.
In this study, patients dropped-out early due to adverse events or progressive disease, and we therefore considered the potential for confounding bias. We examined the study population except for early drop-out cases in which patients discontinued treatment until cycle 2 because of severe adverse events or progressive disease in the same multivariate analysis. In exception cases, total dose until the second cycle <3180 mg (HR 1.97, 95% CI, 1.00–3.86, P = .0496) was extracted as a statistically significant independent poor prognostic factor (Supplementary Table S1). These results clearly demonstrate the clinical significance of the cumulative dose of regorafenib in the early cycles with regard to treatment efficacy in patients with mCRC.
A total of 122 of 176 patients (69.3%) in this study were treated with regorafenib at an initial dose of 160 mg because the study duration ranged from the time regorafenib went on the market to the close of observation. However, the number of patients treated with an initial dose ≤120 mg is currently increasing as a means of preventing discontinuation due to intolerable toxicity. In a recent meta-analysis, treatment with regorafenib at the standard dose of 160 mg was associated with a significant increase in adverse events related to permanent discontinuation, dose interruptions, and dose reductions. 13 Optimizing treatment by means such as personalizing the regorafenib dose and schedule adjustments is common in clinical practice, and many physicians have adopted an empirical approach to manage toxicity as a result of phase III studies. 14 A recent observational cohort study suggested that individualized dosing strategies in patients with mCRC might lead to improved clinical outcomes. 15 In the CORRELATE prospective observational study, the regorafenib toxicity profile was similar to that reported in phase III trials. The starting dose for almost half of the patients in that study was less than the approved 160 mg dose, and the median OS and progression-free survival were in the ranges observed in phase III trials. 16 In the ReDOS study, the dose-escalation group achieved cycle 3 of treatment, but the standard-dose group did not. 7 The results of these studies indicate that optimizing the initial dose is associated with outcome and toxicity, although a relationship between cumulative dose and outcome was not reported. Furthermore, schedule adjustments or discontinuation/restarting, which often occur in real-world settings, were not considered except for the CORRELATE study. Our study shows that cumulative dose until the second cycle in a real-world setting is associated with OS. The association was not statistically significant with the initial dose distribution divided based on median total dose, whereas initial dose was extracted as a prognostic factor in the multivariate analysis. These results indicate that the initial dose should not be decreased arbitrarily and that an individualized starting dose should be considered, consistent with other studies. Although we also examined association relative dose intensity (RDI) until the second cycle with OS, it was not significant by log-rank test (p = .670). On the other hand, we also examined whether initial dose was associated with RDI or not. RDI of the first cycle was statistically significant between 120 mg and 160 mg of initial dose (p = .009), but that of the second cycle was not significant by Mann–Whitney test (p = .135). This result indicated that RDI may be preserved even with early reduced initial dose avoiding severe adverse events.
The respective incidences of HFSR, liver dysfunction, and hypertension were 80%, 31%, and 60% in the Japanese population in the CORRECT study, 4 in contrast to 93.1%, 25.5%, and 35.2%, respectively, in this study. The frequency of hypertension in this study was lower than previously reported, whereas that of HFSR was higher. The rates of adverse events of ≥grade 3 were similar to other studies. In groups separated by median total dose, all grades of HFSR were statistically significant, although the frequency of HFSR was generally over 90% in both groups. These results indicate that HFSR is likely to occur in mCRC patients treated with regorafenib. The data also indicate that the incidences of skin rash and emergency hospitalization in patients with a total dose until the second cycle <3180 mg are clearly higher than in patients in the other group. The results show that skin rash and emergency hospitalization are direct causes of discontinuation or dose reduction. It is therefore important to identify patients who are likely to develop severe adverse effects.
Many researchers have examined ways to optimize the dose of regorafenib, but there are no significant real-world data available. We assessed adherence to regorafenib in order to examine real-world doses. It has not been previously reported that cumulative dose is associated with survival time in view of real-world adherence data. Our study indicates that total dose until the second cycle ≥3180 mg prolongs OS. This value may represent a cut-off point. A regorafenib initial dose of 80 mg continuing until second cycle at the standard schedule would lead to a cumulative dose of 3360 mg in the absence of discontinuation or dose reduction. That is the indicator for regorafenib treatment design in terms of dose-escalation, dose reduction, or schedule adjustment.
Since regorafenib was approved, many studies have examined whether pharmacokinetic and pharmacodynamic parameters such as dose setting are associated with efficacy or adverse events. In general, regorafenib is metabolized by cytochrome P450 3A4 in the liver to its active metabolites, M-2 and M-5. Kubota et al. 17 examined the area under the unbound plasma concentration–time curve (AUCu) for these compounds. Higher AUCu values for M-2 and M-5 on day 1 were associated with significantly shorter progression-free survival than higher AUCu values for total plasma or unchanged drug. Moreover, the RDI during cycle 1 in patients with higher AUCu values for M-2 or M-5 was lower than that for patients with lower AUCu values. These results suggest that the standard dose was too high and that active metabolites played a significant role in patients’ decisions whether to continue treatment. In terms of genetic factors, Kubota et al. reported a significant association between the ABCG2 421 A/C genotype and AUCu values for the active metabolites, whereas another study reported that other genetic factors were not associated with regorafenib pharmacokinetics. 18 Thus, whether genetic factors actually affect regorafenib efficacy and toxicity remains unclear and should be examined in future studies.
There were
Conclusion
The cumulative dose of regorafenib until the second cycle in patients with mCRC is associated with drug efficacy. It is important to determine the optimal regorafenib dose in individual mCRC patients in order to avoid discontinuation or dose reduction, as data regarding regorafenib pharmacokinetics and the effects of genetic factors are inadequate.
Supplemental Material
sj-pdf-1-dos-10.1177_15593258211047658 – Supplemental Material for Association Between Regorafenib Dose and Efficacy Against Metastatic Colorectal Cancer in a Real-World Setting
Supplemental Material, sj-pdf-1-dos-10.1177_15593258211047658 for Association Between Regorafenib Dose and Efficacy Against Metastatic Colorectal Cancer in a Real-World Setting by Masahiro Hatori, Kazuyoshi Kawakami, Takeru Wakatsuki, Eiji Shinozaki, Kazuo Kobayashi, Takeshi Aoyama, Yasuhiro Nakano, Kenichi Suzuki, Kensei Yamaguchi and Toshihiro Hama in Dose-Response
Footnotes
Authors’ Contributions
Conceptualization and study design: MH and KK; writing an original draft: MH; review and editing: KK, TW, KK, ES, and KS; supervision: KY and TH; and investigation of data: MH, KK, TW, KK, TA, and YN. All the authors read and approved the final manuscript.
Declaration of Conflicting Interest
K. Yamaguchi has received speaking honoraria from Taiho Pharmaceutical, Chugai Pharmaceutical, Merk Serono, Takeda Pharmaceutical, Yakult, Bayer, Ono Pharmaceutical, Eli Lilly, Sanofi, and Bristol-Myers Squibb; has received research grants from MSD, Ono Pharmaceutical, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, Daiichi Sankyo, Eli Lilly, Gilead Sciences, and Yakult; and has had a consulting or advisory role for Bristol-Myers Squibb. The other authors declare that they have no conflicts of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.