
Abstract
Metformin pharmacokinetics in a liquid extemporaneous formulation from commercial tablets was determined in paediatric patients. A randomized, transversal clinical trial was conducted in 34 children and adolescents between 7 and 17 years of age. 17 children were randomized to take metformin in the liquid formulation and, after a 1-week wash period, a 500 mg metformin tablet was administered to them. Blood samples were obtained in Whatman 903® cards at 0, 1, 2, 4, 8, 12 and 24 hours. Extraction was made by direct precipitation with acetonitrile (ACN) and methanol, detection by UPLC and tandem mass spectrometry. The method was accurate, precise, selective and linear from 50 to 1000 ng/mL (r = .9982). Comparative pharmacokinetics, tablet vs formulation, were as follows: Cmax 1503.2 ng/mL vs 1521.4, Tmax 1.5 h vs 2.3, and half-life 8.2 vs 7.5 h. The liquid formulation of metformin showed similar pharmacokinetics to the tablet, and the ratios (90% CI) of geometric mean for metformin were 100.63% (89.13–113.6), 98.08% (88.04–109.2), and 97.52% (84.9–112.01), for Cmax, AUC0-t, and AUC 0-∞, respectively. Pharmacokinetics was determined using WinNonlin Pro 3.1 software. The liquid formulation of metformin showed similar pharmacokinetics to the tablet, allowing a more precise dose adjustment and ease of administration.
Introduction
The pharmacological management of obesity and insulin resistance, as a prelude to type II diabetes mellitus (T2DM) and metabolic syndrome (MS) in children and adolescents, has been more frequent due to the failure of exercise and diet therapy,1,2 bad eating habits, stressful lifestyle and prevailing sedentary lifestyle caused by the indiscriminate use of instruments that have made life more comfortable, which has minimized energy expenditure in the most trivial activities of today’s society. 3 Hence, obesity and its related metabolic alterations have recently been installed in pediatrics and, alarmingly, are expected to increase. Metformin hydrochloride is the drug that is used in the first instance in this scenario,4–6 since it helps to reduce the absorption of glucose from the intestine into the blood, inhibits the release of glucose from the liver and makes that skeletal muscle and adipose tissue respond more efficiently to insulin.7,8 Likewise, it helps to restore lipids to normal levels, since it promotes the oxidation of free fatty acids, 8 without causing hypoglycemia or lactic acidosis.
Patients between 12 and 17 years of age who require treatment with metformin can take doses similar to those of adults, depending on their body weight; 9 however, in adolescents without overweight and in those under 8 years who present these same metabolic alterations, it is required to prepare master formulations from the drug in its commercial presentation, according to the specific needs of each patient, such as adjusting dosage, adaptation for swallowing and improvement of taste from commercial tablets. 10
The purpose of the present study was to compare the pharmacokinetics of metformin, in the liquid formulation sweetened without calories, previously developed in our laboratory, 11 with that of the 500 mg tablet in a cohort of 34 paediatric patients who take metformin prescribed by their physician, in order to demonstrate that the concentration of metformin in blood does not change significantly after modifying its presentation, and therefore, it can be used safely in this population.
Materials and Methods
Paediatric Patients
This study included 34 Mexican paediatric patients, children or adolescents, aged 7–17 years, 22 girls and 12 boys, with a diagnosis of overweight, obesity, insulin resistance, diabetes mellitus I or diabetes mellitus II, attended in the Endocrinology Service of the Mexican National Institute of Pediatrics. Informed consent was obtained from all individual participants included in the study. All the patients had metformin prescribed, in monotherapy or combined, or were candidates to receive it and agreed to participate voluntarily in the study, by signing informed consent by their parents or guardians and by the patient when he/she was older than 10 years of age and once the procedure, purpose and advantages of the study had been fully informed.
The patients were out-patients and were never hospitalized. For the study, they stayed in the simple-taking room of the hospital for 12 hours (6
All the patients remained under surveillance in an appropriate consulting room, with food restrictions and without doing any vigorous physical activity, they only had access to board games, check their social networks and play video games on their mobile devices.
The patients who were recruited were asked to eat a light dinner 1 day before the study (approximately 390 kcal). On the day of the study, they were given a standardized diet totalling approximately 2200 kcal, taken in 3 rations as follows: 1 ration for breakfast (approximately 590 kcal), another for lunch (approximately 845 kcal) and the last for dinner (approximately 845 kcal). After the 24-hour sample collection, they were dismissed to return to their houses.
Ethical Aspects
The committees of our institution, Research Committee from the Division of Investigation, National Institute of Paediatrics (17CI09003109), Ethics Committee of the National Institute of Pediatrics (CONBIOETICA-09-CEI-025-20161215) and the Committee of Biosafety in Investigation of the National Institute of Paediatrics (17CB09003143), have given the institutional registration number 045/2016 to the present study in the 23rd of August of 2016. The international registration number is IRB00008064, by the Ethics Committee. This study was carried out with strict adherence to the ethical principles enunciated in the Declaration of Helsinki and to the CONSORT guidelines, http://www.consort-statement.org/consort-2010.
Study Design
Clinical, prospective, longitudinal, analytical, randomized crossed study in 34 paediatric patients. Their treating physician referred paediatric patients, from the Endocrinology Department. Given that they had already prescribed metformin, they were asked to stop taking it from the noonday of the previous day, in order to minimize the basal levels of the pharmacokinetic profile. Seventeen patients were administered a 500 mg tablet of metformin of the innovative brand, with 50 mL of drinking water. The other 17 were given a spoonful (5 mL) of the solution sweetened with metformin (with 1% sucralose), made from the innovative tablets (Glucophage®, Roche®) at a final concentration of 500 mg in 5 mL. The assistance of a maximum of 4 patients per day was scheduled, in order to execute in time and form the taking and handling of the samples.
Throughout the procedure, each patient was comfortable and had medical surveillance, nutritional control and vital signs recording (blood pressure, heart rate, respiratory rate and temperature). All patients were given a hypocaloric breakfast, 2 hours after taking the medication, and their blood glucose levels were monitored. In case of showing signs of hypoglycaemia verified by dextrosticks, a solution of 100 mL with 20% glucose was prepared. In no case was it necessary to administer it.
Study Subjects
Inclusion criteria
Patients with metformin prescribed, in monotherapy or combined, or being candidates to be treated with metformin, for body weight control, insulin resistance or T2DM, who accepted voluntarily to participate after signing the informed consent or assent.
Exclusion criteria
Patients with drug abuse or addiction background were excluded from the study, as well as those with renal or hepatic failure, adolescents with venereal diseases, hepatitis or being HIV positive, and also the girls with confirmed pregnancy or lactation, and those whose clinical condition is caused by a genetic syndrome or pharmacologic treatment or individuals who received a blood transfusion in the last 3 months.
Elimination criteria
Patients with incomplete number of samples or in deficient amounts or qualities to quantitate metformin were eliminated from the study and also those who decided to voluntarily quit the study in any stage. Treatment-naïve patients, hypersensible to biguanides, were initially considered to be eliminated in case of adverse reactions; however, this situation did not occur in any of the cases.
Sample Size
It was determined to employ 36 individuals to determine the complete pharmacokinetic profile of 24 hours, according to international standards of studies of bioavailability and bioequivalence. Two of them were considered under elimination criteria, because they did not complete the first stage of the study and did not attend the second stage, so that their number of samples was incomplete.
Random Allocation
We carried out the randomization, both in the administration of the tablet and the solution, as well as in assigning the stage of administration, that is, in determining which pharmaceutical form was going to be administered first – the tablet or the solution.
The Excel program was used to generate random numbers. Of these numbers, the first 17 numbers generated were marked and later the cell was copied and ordered from smallest to largest. Those that were marked were assigned to the solution and those that were not, were assigned to the Tablet.
On the other hand, random numbers were again generated to assign the administration of tablet or solution to the first phase of the study or to the second phase. The same procedure described above was followed.
Blood Samples Collection
Seven blood samples were obtained from each patient by digital puncture with contact-activated lancets BD Microtainer at 0, 1, 2, 4, 8, 12 and 24 hours after taking the metformin tablet (500 mg) or the sweetened formulation (500 mg/5 mL). These samples were collected as drops on Güthrie cards and stored in airtight plastic bags with desiccant, at a temperature of 2–8°C until the moment of their analysis.
Reagents
Pure standards of metformin hydrochloride and ranitidine were purchased from MP Biomedicals (Fountain Pkwy, Solon OH, USA®). Acetonitrile and HPLC grade methanol from JT Baker. Ammonium acetate was from Merck (Darmstadt, Germany®) and formic acid from Sigma Aldrich® (St. Louis MO, USA). For all solutions and dilutions, bidistilled water filtered by Milli-Q system (Millipore, Molsheim, France®) was used. The solvents used were mass grade.
Chromatographic Conditions
We used an ultra-high performance liquid chromatography system, AcQuity model (Waters Co., Milford MA, USA®) coupled to a Quattro-micro tandem mass spectrometer (WatersMicromass, Manchester, UK®), operated in the ionization mode, positive by electronic spray (ESI (+): ‘electrospray positive ionization mode’) and controlled by the MassLynx NT 4.0 computer program (WatersMicromass, Beverly, MA, USA®). The separation of the compounds was carried out on an Acquity UPLC BEH-HILIC column, 2.1 × 100 mm, 1.7 μm (WatersTM) at 40°C, autosampler at 15°C, flow rate of .25 mL/minutes. Running time was 3.5 minutes. The retention times were 2.07 minutes for metformin and 2.51 minutes for ranitidine. The mobile phase consisted of .5 M ammonium acetate, with acetonitrile (ACN), in 80:20 proportions, respectively.
Spectrometric Conditions
The analytes were measured by multiple reaction monitoring, using the following ion transitions: m/z1 + 130> 70.58 Th for metformin and 315.17 > 176.05 Th for ranitidine (internal standard). The cone energy was 20 and 25 V, for metformin and ranitidine, respectively, while the collision energy was 20 V for both. A dwell of .25 s was used.
The MassLynx Ver. 4.1 software processed the entire data. The separation of the metformin and ranitidine (internal standard) was carried out on an Acquity UPLC BEH-HILIC column, 2.1 × 100 mm, 1.7 μm (WatersTM) at 40°C, autosampler at 15°C, flow rate of .25 mL/minute. Running time was 3.5 minutes.
Standards and Quality Controls
Initial solutions of metformin and ranitidine (1 mg/mL) were prepared individually in 50% methanol (v/v, in water). From the initial solution, dilutions were made in 50% methanol to obtain working solutions. These solutions were used to prepare the calibration curves and control points with final concentrations of 50, 200, 400, 600, 800 and 1000 ng/mL, according to the plasma concentrations reported in Hispanic adolescents. 12 The low, medium and high control points were 150, 500 and 750 ng/mL. The calibration curve was prepared by mixing 950 μL of whole blood (haematocrit 45%) with 50 μL of metformin working solution. 40 μL of this mixture was dripped onto Whatman S & S 903 cards (Güthrie cards) in each circle separately. The cards were allowed to dry horizontally for 12 hours at room temperature. Once dried, they were stored appropriately labelled in plastic bags with low gas permeability, accompanied by drying material, at −80°C until analyzed.
Sample Processing (Extraction)
Five 3.0 mm diameter discs were cut from each sample circle on the card, using a McGill, Incorporated® hand drill. The appropriate number of discs was placed in a 1.5 mL microtube, and 750 μL of a mixture of methanol and ACN was added in a 67:33 v/v ratio. They were stirred for 2 minutes at room temperature on a tap shaker (Vortex®) and then placed in a bath with ultrasound for 5 minutes. The samples were centrifuged at 12000 rpm for 5 minutes at room temperature. The organic phase was collected and transferred to another tube, evaporated at 40°C under a stream of nitrogen. The extracted and dried sample was reconstituted with 150 μL of a ranitidine solution of 25 ng/mL in water: ACN, in proportion 70:30 (v/v). For detection, 5 μL were injected into the chromatographic system.
Method Validation
The validation of the present method was carried out with strict adherence to the official Mexican norms NOM-177-SSA1-2013 (FDA 2001, EMEA), and in turn, in accordance with international guidelines for bioanalytical methods. The tests performed in UPLC-MS/MS were: ion suppression by matrix effect, selectivity of the method for concomitant drugs (amoxicillin, acetylsalicylic acid, captopril, orlistat, omeprazole and acetaminophen), carry over, linearity, precision, accuracy, lower limit of quantification (LLOQ), absolute recovery and stability of the drug in solution and extracted from the sample in the short and long term.
Pharmacokinetic Parameters
The pharmacokinetics of metformin in extemporaneous formulation, compared with that of the tablet, was characterized. The parameters calculated were the maximum plasma concentration (Cmax), the time in which it is reached (Tmax), the elimination half-life (t1/2), the elimination constant (ke), the apparent volume of distribution (Vd), the clearance (Cl), the area under the curve (AUC0-12) and the area under the curve from time zero to infinity (AUC∞). The average results of all patients are presented, including the standard deviation (Table 2).
Statistics
The analysis of variance is carried out considering the following sources of variation:
Si (Gj) = Subject nested in the sequence, Gj = Sequence of administration, Pk = Period, Fj, k = Formulation
The analysis was performed using the mixed-effect model, leaving the sequence, product and period sources of variation as fixed effects and the subject factor nested in sequence as random effects. In the analysis of variance, values of P > .05 are observed for the sources of variation formulation, sequence and period, reason why it was concluded the existence of a non-significant effect of the sources of variation formulation, period of administration and sequence of administration. With the latter, the presence of a carry-effect is ruled out.
On the other hand, the inter-individual variability observed in terms of CV% was 57%, 69.09% and 88.42% for Cmax, AUC0-t and AUC0-inf, respectively. In addition, the residual variability, also known as within-subject variability, was 29.71%, 26.56% and 29.83% in terms of CV% for Cmax, AUC0-t and AUC0-inf, respectively.
Results
Demographic Data of Patients in the Cross-Study.

Note: Stage I: From day 1 to day 30, group 1 received metformin in liquid formulation; group 2 received the fractionated tablet. Stage II: From days 37 to 67, the pharmaceutical presentations of metformin that both groups took were invested.
Abbreviation: BMI, body mass index.
As shown in Figure 1, the pharmacokinetic profiles of metformin in the liquid formulation and in the tablet had practically the same behaviour in the paediatric population analyzed. The pharmacokinetic parameters of metformin in children and adolescents are presented in Table 2. At a dose of 500 mg, the tablet produced a maximum plasma concentration (Cmax) of 1503.2 ng/mL, which was practically the same value as with the liquid formulation (1521.4 ng/mL); the time to reach the maximum plasma concentration (Tmax) of the tablet was 2.3 h, compared with that of the solution, which was 1.5 h, without significant differences. Also, the elimination constant (ke) was .1 h−1 for the tablet, whereas it was .08 h−1 with the solution. These data suggest that metformin has the same pharmacokinetic behaviour in the liquid formulation as in the tablet. On the other hand, the apparent volume of distribution was slightly higher in the tablet than in the liquid formulation (Vd = .61 L vs Vd = .46 L), but the difference is not significant. Pharmacokinetic profile of metformin in paediatric patients (n = 34). Average from all the patients after taking a 500 mg dose orally. Compared Metformin Pharmacokinetics. *Statistics were applied on decimal logarithm-transformed data: n = 34. In the pharmacokinetics parameters, values are given as mean ± standard error. Abbreviations: Cmax, maximum plasma concentration; AUC0-12, AUC to last measurable concentration; AUC0-∞, AUC extrapolated to infinity; AUC, area under the curve; IC, confidence interval.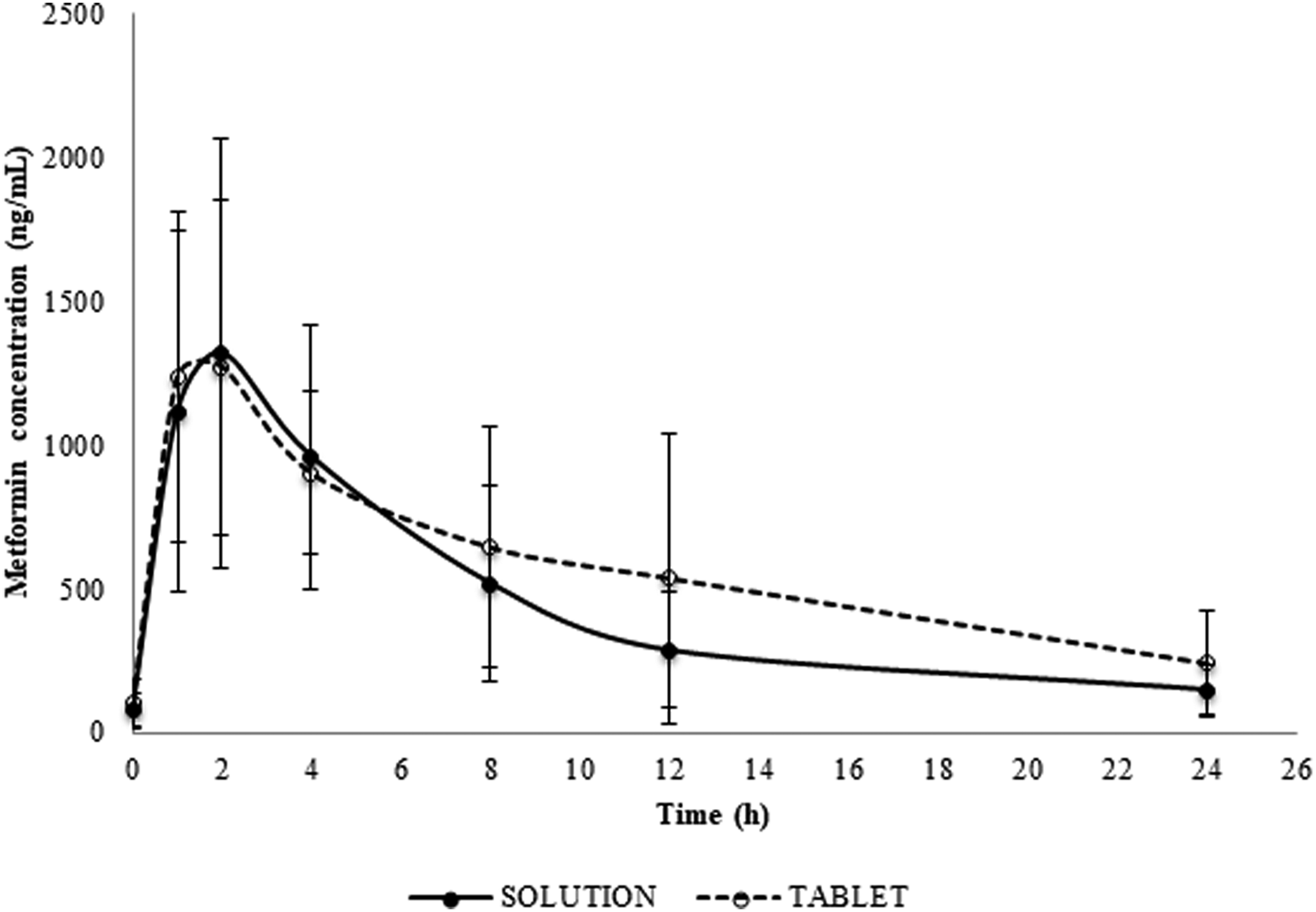

Validation of the Analytical Method
For validation, next parameters were referring
Carry-over: Carry-over was assessed by injecting blank samples, one previous blank sample and two samples after the highest calibration standard. The acceptance criterion was carry-over of less than 20% of the LLOQ and 5% for the IS (Table 3).
Parameter used for Validation of the Analytical Method.

Abbreviations: QC, quality control; ND, not determined.
Recovery was made by comparing the response (peak area) of dried blood spots (DBS) QC (low, mid and high) sample extracts and blank DBS sample extracts post-fortified with the same DBS QC concentration. The recovery was 96.1%, 99.5% and 99.1% for low, mid and high-quality control, respectively, whereas for the internal standard, it was 100.4%.
Haematocrit effect: Blood samples were prepared with different percentages of haematocrit (35–50%), each sample containing 650 ng/mL of metformin. The blood samples were processed in triplicate and quantified using a calibration curve prepared with 40% haematocrit. This value was selected according to Lopez Santiago, 13 who indicated that children from 6 to 12 years old have a haematocrit of 40%, while male adolescents from 12 to 18 years old have a haematocrit of 43%, and for women, it is 41%.
Effect of Haematocrit on the Quantification of Metformin (650 ng/mL).

The matrix effect was determined by extracting blank samples in DBS and, at the end of the process, solutions of the analyte and the internal standard (quality controls low quality control, median quality control and high quality control) were added and their responses were separately inter-compared. For each unit, a normalized matrix factor (NMF) was obtained, according to the following formula.
Matrix blank samples were extracted, which after extraction were reconstituted with metformin and ranitidine in solution to obtain the corresponding analytical response. The analytical response was compared with the analytical response obtained from the analyte and the IS in solution. The matrix effect was determined according to the following formula.
Normalized Matrix Factor: NMF = (Response of the analyte in the matrix/response of the internal standard in the matrix).
(Response of the analyte in solution/response of the internal standard in solution).
The acceptance criterion for the matrix effect indicates a coefficient of variation of less than 15% for the NMF. In our trial, values of 4.43%, 9.31% and 3.7% of CV were obtained for the concentrations of the quality controls, low, medium and high, respectively. Our results indicate we did not find matrix effect.
Selectivity: The method was selective at concomitant drugs (amoxicillin, acetylsalicylic acid (Aspirin), captopril, orlistat, omeprazole and acetaminophen).
The method developed and validated in this work for the quantification of plasma levels of metformin proved to be linear in the range of concentrations from 50 to 1000 ng/mL. A coefficient of determination (r2) of .998035 was obtained. The intra- and inter-day variability met the acceptance criteria, and the method was precise and accurate, with coefficients of variation of less than 7.5%. The power obtained in the study was 84% for Cmax, for AUC0-t 89% and for AUC0-inf it was 82% (Table 2). The samples extracted and stored at 4°C were stable for 17 hours and also stable up to 15 hours in autosampler (15°C) and up to 2 months at −80°C.
Spectrometric Conditions for Metformin Detection by UPLC-MS/MS.

Note: Ranitidine was used as internal standard.
Abbreviations: V, volt, kV, kilovolt.
Discussion
Currently, no studies of bioavailability of liquid metformin have been reported in paediatric patients. In fact, in several Latin American countries, a paediatric presentation of metformin has not been put on the market because this population constitutes a much smaller market than that of adults; it requires smaller volumes of product and with a greater variety of doses, due to weight and height variations in this age group.14,15 In most cases, medical staff and family members must split the tablets to adjust the dose. This procedure temporarily and partially solves the problem of giving the medication to the child; however, it may cause sub-therapeutic doses, difficulty administering the medication or a plasma concentration that is not constant.
In the present study, the determination of the relative bioavailability of metformin in liquid formulation, sweetened with 1% sucralose, which is a non-caloric sweetener, was carried out. The extemporaneous metformin solution was made from Roche Glucophage® tablets, containing 500 mg; bottled drinking water was used for the vehicle of the formula, and the bitter taste was masked by adding commercial Splenda® sweetener (sucralose, Johnson & Johnson) without calories. It was evaluated for 30 days at 25 ± 2° C, exposed to light and darkness, 4°C and 40°C. We found that it was stable under all storage conditions. 16 In previous studies, it has been shown that this formulation, developed in our laboratory, is stable physicochemically for up to 30 days at room temperature and up to 60 days in refrigeration; likewise, it showed microbiological stability, since no growth of aerobic mesophilic microorganisms, enterobacteria or fungi was observed. 17 It is advisable to evaluate the bioavailability of extemporaneous formulations in healthy adult volunteers and extrapolate the results to the paediatric population, since it is not ethical to perform this type of study in healthy children.
Due to the fact that a study was conducted in healthy volunteers of the extemporaneous liquid formulation elaborated in our laboratory, obtaining no reaction or adverse effect to metformin in this liquid presentation, 18 and in view of the need for a pharmaceutical presentation for administration in paediatric patients, it was decided to perform this bioavailability study in a paediatric population. With this, it has been demonstrated that there are no significant differences between the pharmacokinetic parameters of the formulation, compared with those of the commercial tablet in this population. Likewise, there were no adverse reactions to the extemporaneous formulation of metformin, which suggests that the liquid formulation that we have evaluated can be administered safely in the paediatric population. 19
The solution has a bioavailability similar to that of the tablet; therefore, it can be used in paediatric patients.
On the other hand, in our study, we had a difference in terms of the half-life obtained and that reported in the literature. In our study, the half-life time ranges from 3 to 12 hours, when it is reported to be around 6.5 hours. The fact that the half-life was reached at about 12 hours in the children is probably due to the characterization of the phase of elimination, which might have been affected by the restriction to take more samples from patients to characterize such elimination. Additionally, our study was carried out in patients with T2DM, unlike other studies that were carried out in healthy adult volunteers. In the study performed by Sanchez et al in 2011, 12 to characterize the pharmacokinetics of metformin in 69-year-old, non-diabetic or obese girls, the authors obtained a half-life time of 4 hours. However, in our study, all the participants had T2DM; therefore, the clinical condition of the patients could be the cause of the difference in the half-life time. On the other hand, it is possible that there is a genetic variant in the Mexican population, which makes the renal transport of metformin to be incorrectly expressed, the rs117483482/A gene in MATE2 (metformin transporter encoding MATE2). MATE2 is a protein found in the kidney for the expression of metformin. Therefore, it is likely that this transporter does not act efficiently, thus making the drug to accumulate and increasing its half-life time. However, studies on polymorphisms are required to demonstrate this speculation.
The liquid formulation of metformin, developed in our laboratory, contains 10% sucralose, which gave it a sweet and very pleasant taste. However, in the present study, it was decided to reduce its concentration to the minimum required to mask the bad taste of the drug, in order to avoid any possible toxicity caused by the sweetener. 20 Thus, the liquid formulation administered to patients had only 1% sucralose.
In this regard, there are reports that support the safety and innocuousness of sucralose consumption in paediatric patients and in the general population. Studies of the consumption of sucralose in humans, infants, children, adults, pregnant women, lactating women, diabetics and obese have shown that the daily intake of sucralose triples the maximum dose for 3 months, does not produce adverse metabolic reactions, modifies plasma insulin levels or glycaemia levels when ingested in isolation, does not alter glycosylated haemoglobin levels in the long term, does not cause toxicity even with the consumption of 18 kg accumulated during life, and does not modify the expression or function of GLUT-2 receptors, so it does not interfere with the intestinal absorption of glucose. 21 The study carried out to evaluate the bioavailability in paediatric patients included the design of this formulation using 1% sucralose, in order to significantly improve palatability and ensure acceptance of the dose and adherence to its consumption by part of the paediatric population. The use of metformin in paediatrics is approved by the Food and Drug Administration (FDA) for the treatment of diabetes mellitus type II, and its prescription is conditioned in this population when diet and exercise are not enough to reverse insulin resistance, or do not help to lower weight to patients at the appropriate time, according to meta-analysis of non-randomized studies of small groups (cases and controls, cohort, observational).22-24 That is why, in this work was included the bioavailability of the solution developed in our laboratory at the same dose that is prescribed to children and adolescents, according to their requirements for glycaemic control and body weight. It has been demonstrated that the sweetened solution has practically the same pharmacokinetic profile as the tablet so that it can be used in this population.
On the other hand, the determination of plasma levels of drugs is necessary to monitor the adherence to treatment and correlate the therapeutic success. This activity is especially important in the case of drugs adapted to the paediatric population, such as the extemporaneous formulation of metformin. As described above, in this study, we used the dry blood spot technique that is less invasive than venepuncture, to monitor blood levels of metformin. 25 Given that there are no commercial presentations with doses that can be used in paediatric patients, with the exception of EEUU and Canada, where they are already commercialized.26–31
The limitation of our study is that a large number of samples are required to perform the complete characterization of metformin (.0, .5, 1.0, 1.5, 1.75, 2.0, 2.25, 2.5, 2.75, 3, 4, 5, 6, 8, 12, 24 and 36 hours). However, when dealing with paediatric patients, it is difficult to obtain a large number of samples. Another limitation is that the studies carried out in patients are more difficult to do, since they cannot be stopped from administering their treatment, in order not to put their health at risk. Above all, this was previously agreed with the doctor in charge of them. In addition, in studies of this type, a washout period of 1 week should be allowed; this was not carried out since patients cannot be suspended or interfered with the research treatment.
The liquid extemporaneous formulation of metformin used in this work has shown to have a pharmacokinetics similar to that of the tablet in children and adolescents who are prescribed this drug. This suggests that the oral bioavailability of metformin, in the extemporaneous liquid formulation we have proposed, is practically the same as that of the drug in its original form, only that it offers the advantage of having smaller doses than the tablet and content uniformity.
Conclusions
Our extemporaneous metformin liquid formulation has bioavailability comparable to that of the tablet. It is equally bioavailable as the original pharmaceutical form, with the advantage of allowing lower doses than the tablet, with precision, uniformity of content and ease of administration to the paediatric population.
Footnotes
Acknowledgements
We thank Dr Cyril Ndidi Nwoye Nnamezie, an expert translator whose native language is English, for his help in preparing this manuscript.
Author Contributions
All authors meet the following criteria for authorship: (i) made a substantial contribution to the concept or design of the work; or acquisition, analysis or interpretation of data, (ii) drafted the article or revised it critically for important intellectual content, and (iii) approved the version to be published. (iv) All author participated sufficiently in the work to take public responsibility for appropriate portions of the content.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors express their profound gratitude to the National Institute of Pediatrics (NIP) for the support in the publication of this article, derived from the project 045/2016 of the Program A022.